

Processing and turnover of the Hedgehog protein in the endoplasmic reticulum
- PMID: 21357747
- PMCID: PMC3051819
- DOI: 10.1083/jcb.201008090
Processing and turnover of the Hedgehog protein in the endoplasmic reticulum
Abstract
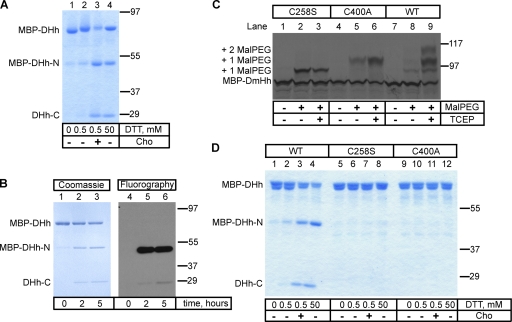
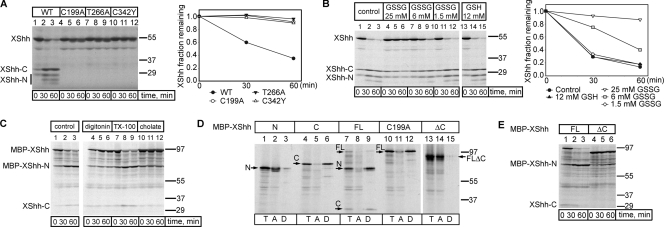
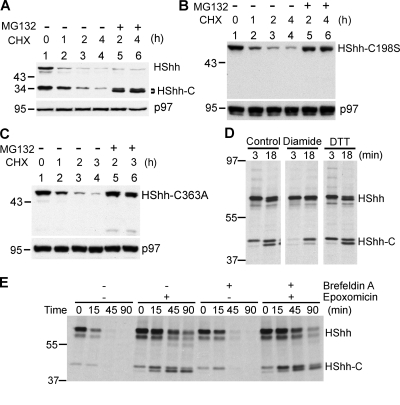
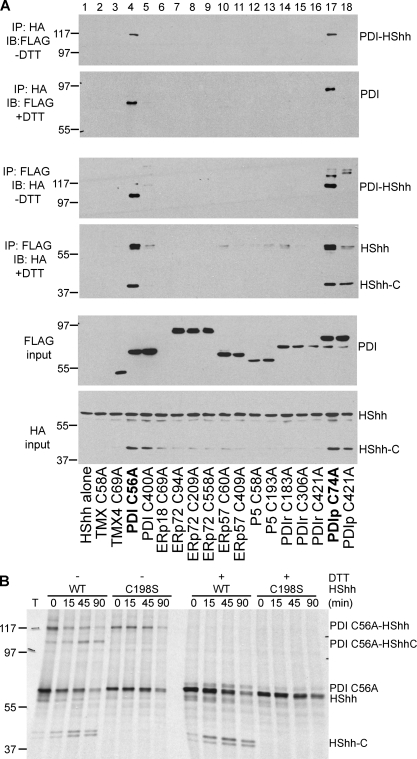
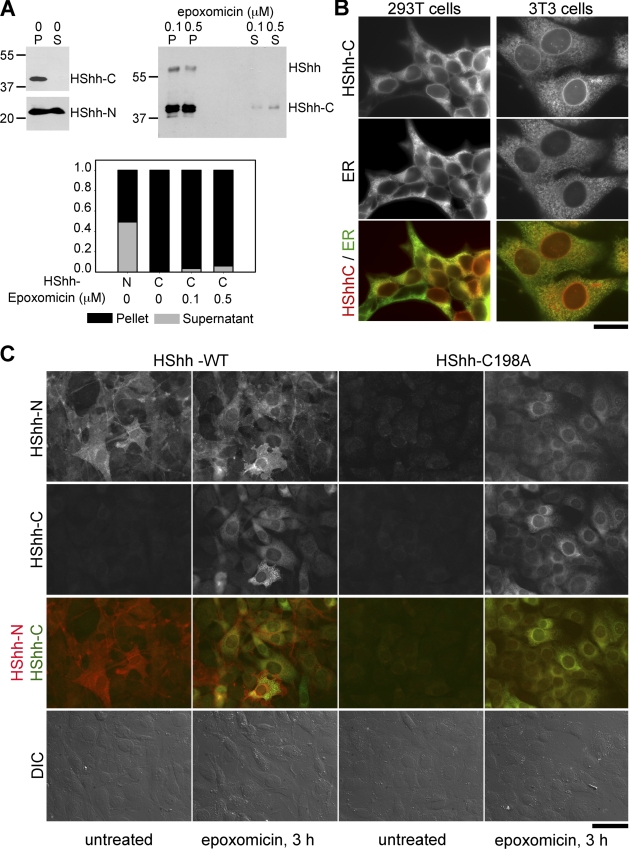
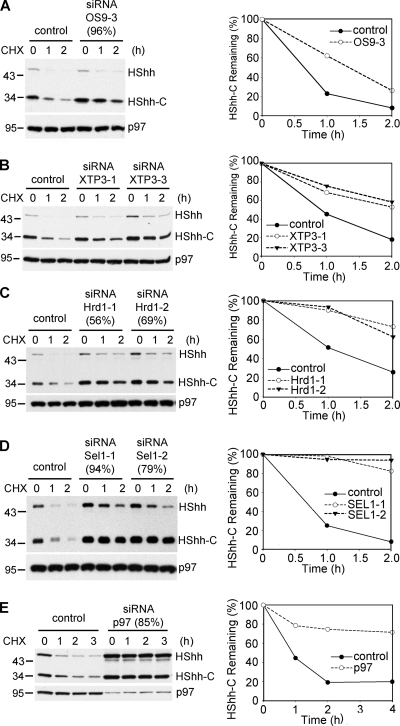
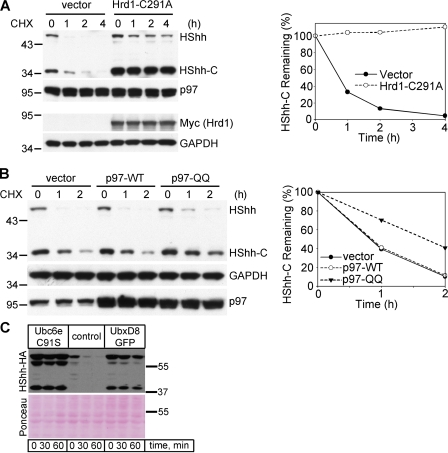
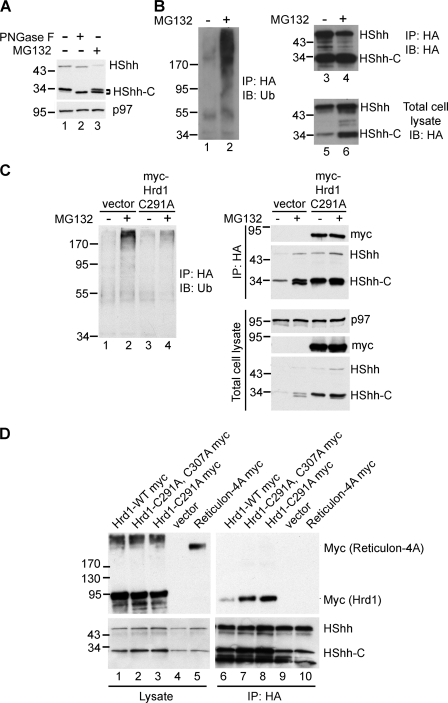
The Hedgehog (Hh) signaling pathway has important functions during metazoan development. The Hh ligand is generated from a precursor by self-cleavage, which requires a free cysteine in the C-terminal part of the protein and results in the production of the cholesterol-modified ligand and a C-terminal fragment. In this paper, we demonstrate that these reactions occur in the endoplasmic reticulum (ER). The catalytic cysteine needs to form a disulfide bridge with a conserved cysteine, which is subsequently reduced by protein disulfide isomerase. Generation of the C-terminal fragment is followed by its ER-associated degradation (ERAD), providing the first example of an endogenous luminal ERAD substrate that is constitutively degraded. This process requires the ubiquitin ligase Hrd1, its partner Sel1, the cytosolic adenosine triphosphatase p97, and degradation by the proteasome. Processing-defective mutants of Hh are degraded by the same ERAD components. Thus, processing of the Hh precursor competes with its rapid degradation, explaining the impaired Hh signaling of processing-defective mutants, such as those causing human holoprosencephaly.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
