

Genome structural variation discovery and genotyping
- PMID: 21358748
- PMCID: PMC4108431
- DOI: 10.1038/nrg2958
Genome structural variation discovery and genotyping
Abstract
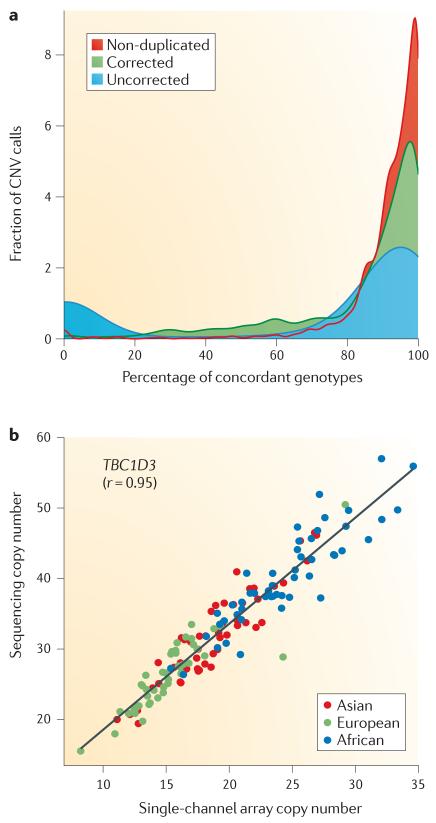
Comparisons of human genomes show that more base pairs are altered as a result of structural variation - including copy number variation - than as a result of point mutations. Here we review advances and challenges in the discovery and genotyping of structural variation. The recent application of massively parallel sequencing methods has complemented microarray-based methods and has led to an exponential increase in the discovery of smaller structural-variation events. Some global discovery biases remain, but the integration of experimental and computational approaches is proving fruitful for accurate characterization of the copy, content and structure of variable regions. We argue that the long-term goal should be routine, cost-effective and high quality de novo assembly of human genomes to comprehensively assess all classes of structural variation.
Figures
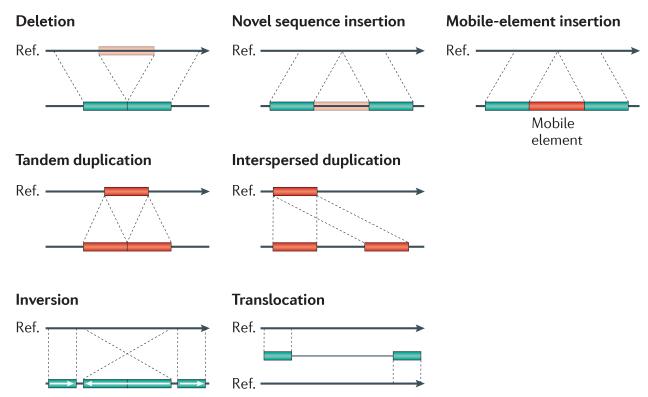
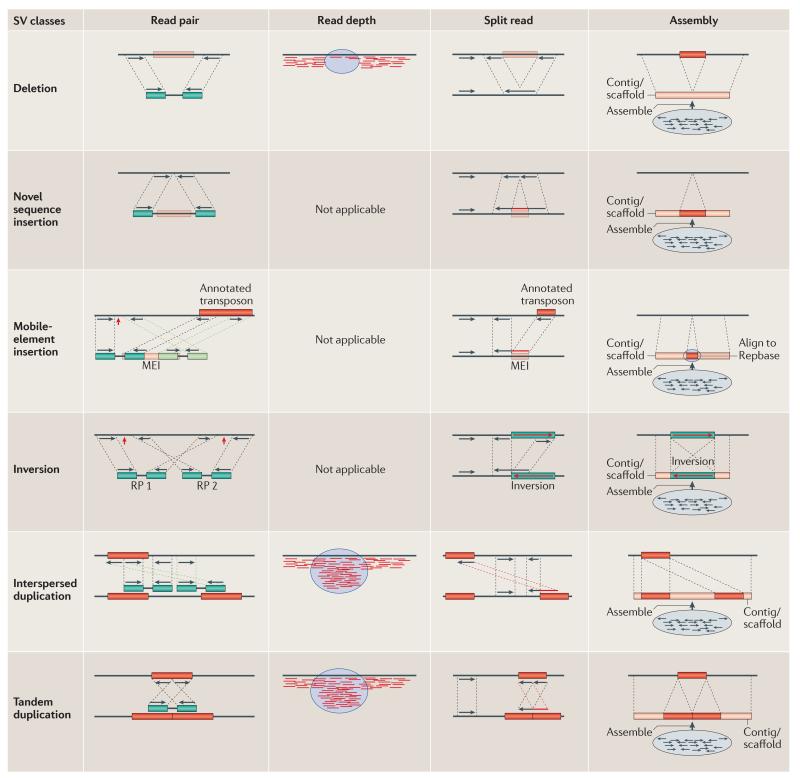
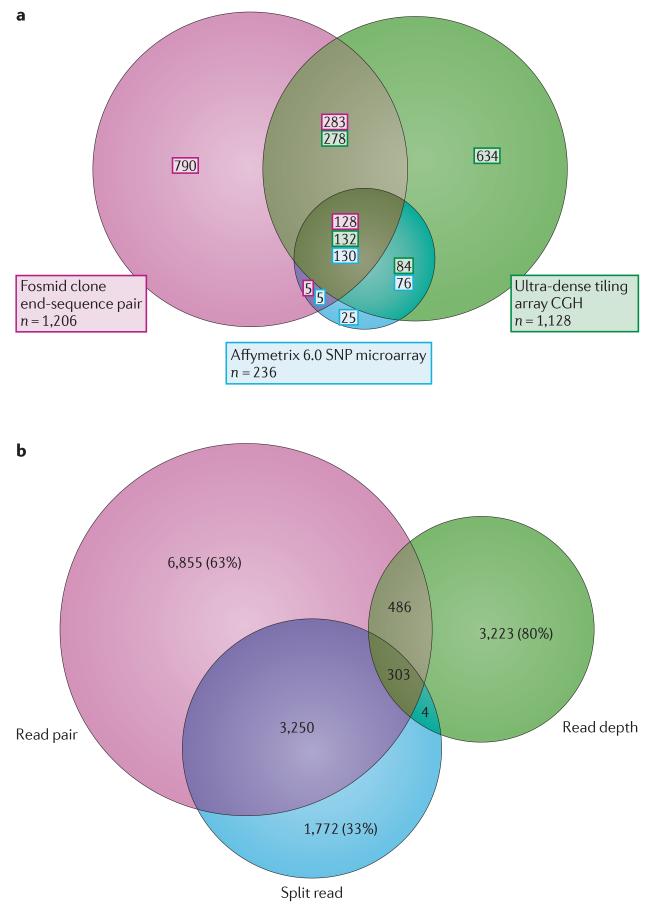
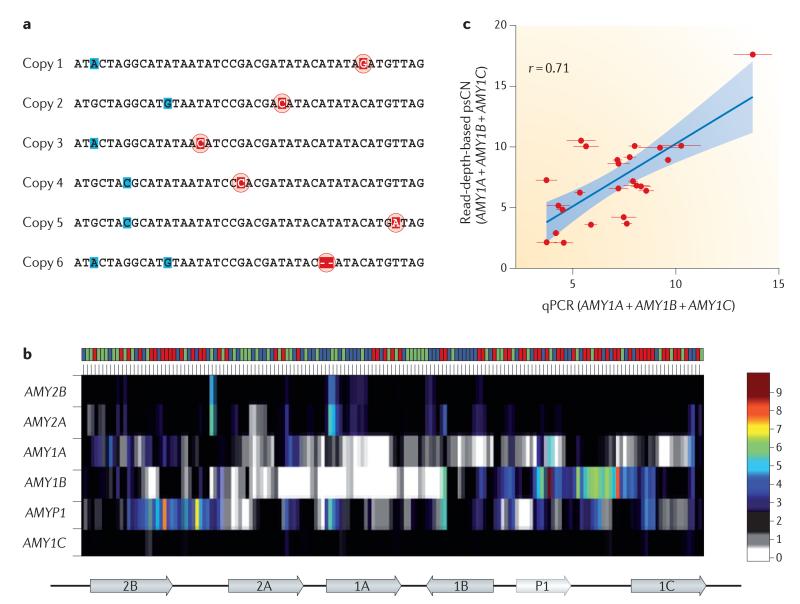
References
-
-
Iafrate A J, et al. Detection of large-scale variation in the human genome. Nature Genet. 2004;36:949–951. The first report of CNVs in the human genome using array CGH.
-
-
-
Tuzun E, et al. Fine-scale structural variation of the human genome. Nature Genet. 2005;37:727–732. The first study to implement a paired-end sequencing approach to study structural variation.
-
-
-
Conrad D F, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. This study represents the first application of an ultra-high-density CGH array.
-
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
