

Microdeletion/microduplication of proximal 15q11.2 between BP1 and BP2: a susceptibility region for neurological dysfunction including developmental and language delay
- PMID: 21359847
- PMCID: PMC6814187
- DOI: 10.1007/s00439-011-0970-4
Microdeletion/microduplication of proximal 15q11.2 between BP1 and BP2: a susceptibility region for neurological dysfunction including developmental and language delay
Abstract
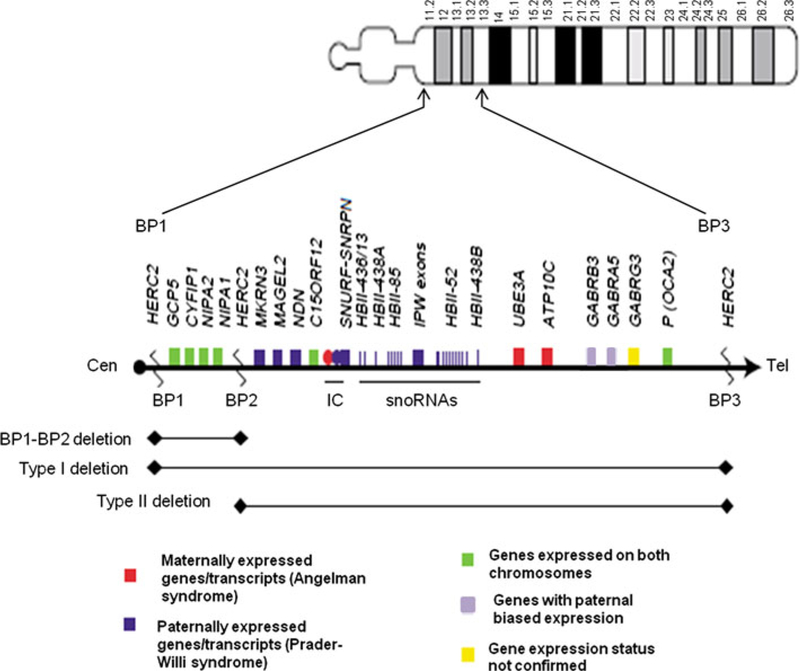
The proximal long arm of chromosome 15 has segmental duplications located at breakpoints BP1-BP5 that mediate the generation of NAHR-related microdeletions and microduplications. The classical Prader-Willi/Angelman syndrome deletion is flanked by either of the proximal BP1 or BP2 breakpoints and the distal BP3 breakpoint. The larger Type I deletions are flanked by BP1 and BP3 in both Prader-Willi and Angelman syndrome subjects. Those with this deletion are reported to have a more severe phenotype than individuals with either Type II deletions (BP2-BP3) or uniparental disomy 15. The BP1-BP2 region spans approximately 500 kb and contains four evolutionarily conserved genes that are not imprinted. Reports of mutations or disturbed expression of these genes appear to impact behavioral and neurological function in affected individuals. Recently, reports of deletions and duplications flanked by BP1 and BP2 suggest an association with speech and motor delays, behavioral problems, seizures, and autism. We present a large cohort of subjects with copy number alteration of BP1 to BP2 with common phenotypic features. These include autism, developmental delay, motor and language delays, and behavioral problems, which were present in both cytogenetic groups. Parental studies demonstrated phenotypically normal carriers in several instances, and mildly affected carriers in others, complicating phenotypic association and/or causality. Possible explanations for these results include reduced penetrance, altered gene dosage on a particular genetic background, or a susceptibility region as reported for other areas of the genome implicated in autism and behavior disturbances.
Figures
References
-
- Antonacci F, Kidd JM, Marques-Bonet T, Teague B, Ventura M, Girirajan S, Alkan C, Campbell CD, Vives L, Malig M, Rosenfeld JA, Ballif BC, Shaffer LG, Graves TA, Wilson RK, Schwartz DC, Eichler EE (2010) A large and complex structural polymorphism at 16p12.1 underlies microdeletion disease risk. Nat Genet 42:745–750 - PMC - PubMed
-
- Baldwin EL, Lee JY, Blake DM, Bunke BP, Alexander CR, Kogan AL, Ledbetter DH, Martin CL (2008) Enhanced detection of clinically relevant genomic imbalances using a targeted plus whole genome oligonucleotide microarray. Genet Med 10:415–429 - PubMed
-
- Blackwell J (2001) Clinical practice guideline: screening and diagnosing autism. J Am Acad Nurse Pract 13:534–536 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
