

Colonization-induced host-gut microbial metabolic interaction
- PMID: 21363910
- PMCID: PMC3045766
- DOI: 10.1128/mBio.00271-10
Colonization-induced host-gut microbial metabolic interaction
Abstract
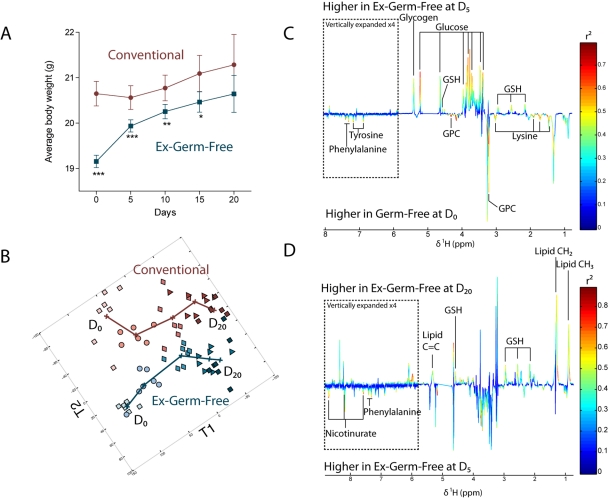
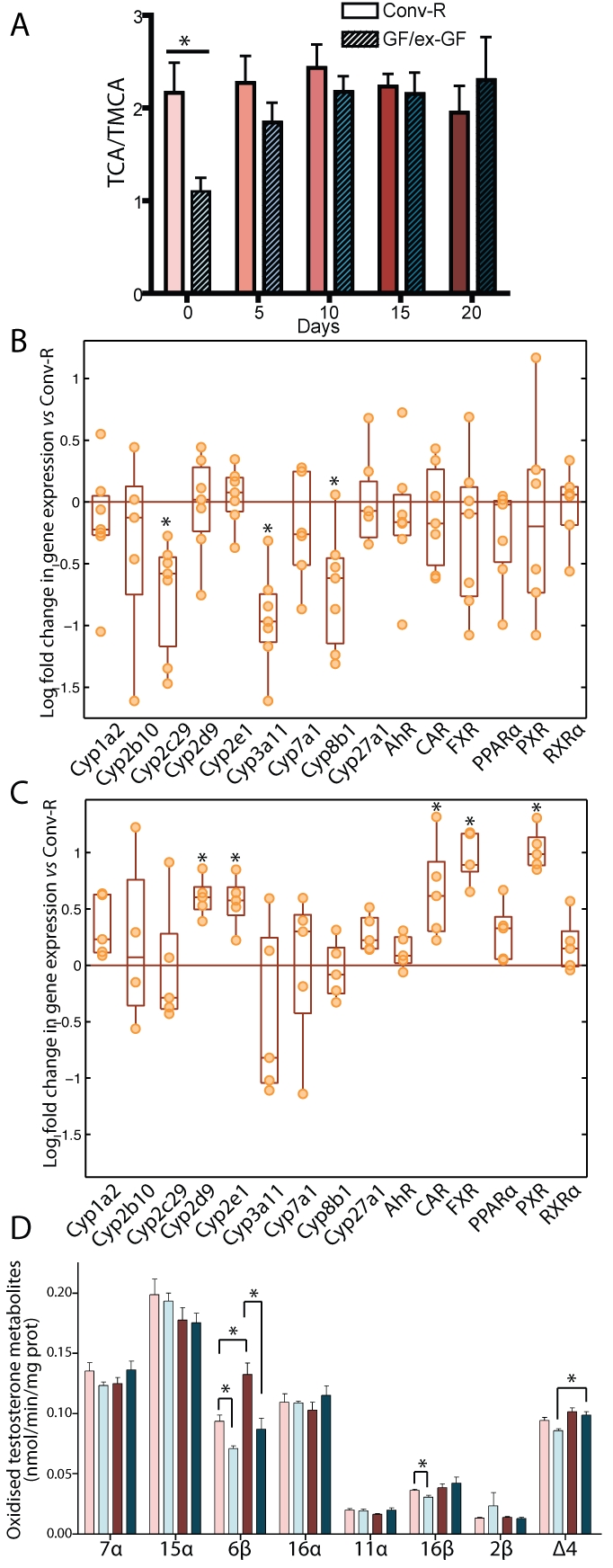
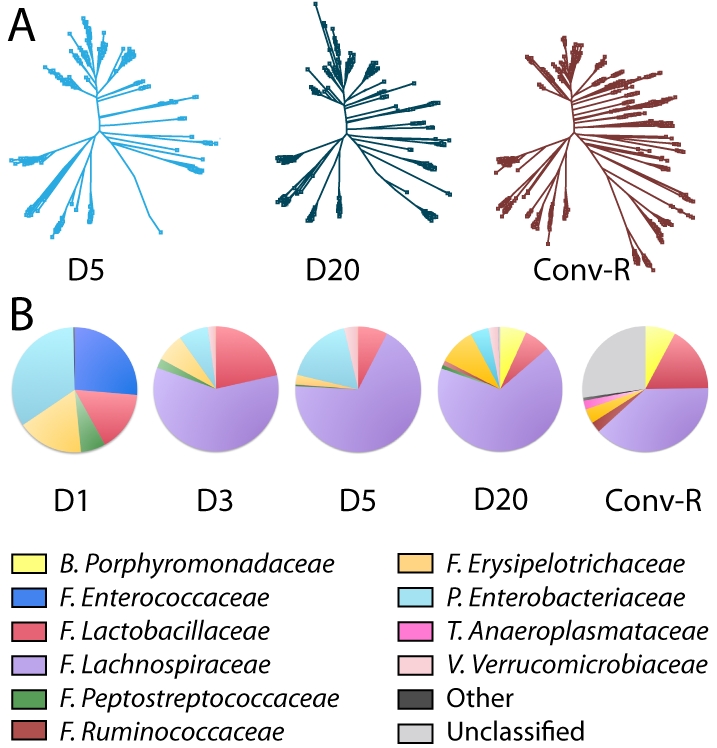
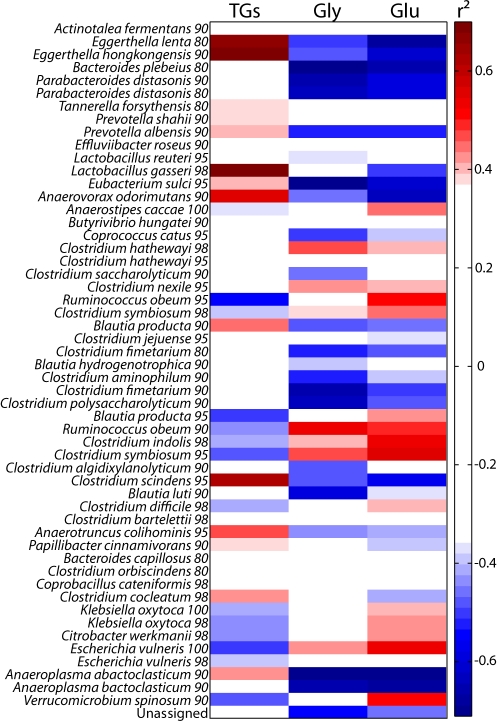
The gut microbiota enhances the host's metabolic capacity for processing nutrients and drugs and modulate the activities of multiple pathways in a variety of organ systems. We have probed the systemic metabolic adaptation to gut colonization for 20 days following exposure of axenic mice (n = 35) to a typical environmental microbial background using high-resolution (1)H nuclear magnetic resonance (NMR) spectroscopy to analyze urine, plasma, liver, kidney, and colon (5 time points) metabolic profiles. Acquisition of the gut microbiota was associated with rapid increase in body weight (4%) over the first 5 days of colonization with parallel changes in multiple pathways in all compartments analyzed. The colonization process stimulated glycogenesis in the liver prior to triggering increases in hepatic triglyceride synthesis. These changes were associated with modifications of hepatic Cyp8b1 expression and the subsequent alteration of bile acid metabolites, including taurocholate and tauromuricholate, which are essential regulators of lipid absorption. Expression and activity of major drug-metabolizing enzymes (Cyp3a11 and Cyp2c29) were also significantly stimulated. Remarkably, statistical modeling of the interactions between hepatic metabolic profiles and microbial composition analyzed by 16S rRNA gene pyrosequencing revealed strong associations of the Coriobacteriaceae family with both the hepatic triglyceride, glucose, and glycogen levels and the metabolism of xenobiotics. These data demonstrate the importance of microbial activity in metabolic phenotype development, indicating that microbiota manipulation is a useful tool for beneficially modulating xenobiotic metabolism and pharmacokinetics in personalized health care.
Importance: Gut bacteria have been associated with various essential biological functions in humans such as energy harvest and regulation of blood pressure. Furthermore, gut microbial colonization occurs after birth in parallel with other critical processes such as immune and cognitive development. Thus, it is essential to understand the bidirectional interaction between the host metabolism and its symbionts. Here, we describe the first evidence of an in vivo association between a family of bacteria and hepatic lipid metabolism. These results provide new insights into the fundamental mechanisms that regulate host-gut microbiota interactions and are thus of wide interest to microbiological, nutrition, metabolic, systems biology, and pharmaceutical research communities. This work will also contribute to developing novel strategies in the alteration of host-gut microbiota relationships which can in turn beneficially modulate the host metabolism.
Figures
References
-
- Ley R. E., Turnbaugh P. J., Klein S., Gordon J. I. 2006. Microbial ecology: human gut microbes associated with obesity. Nature 444:1022–1023 - PubMed
-
- Nicholls A. W., Mortishire-Smith R. J., Nicholson J. K. 2003. NMR spectroscopic-based metabonomic studies of urinary metabolite variation in acclimatizing germ-free rats. Chem. Res. Toxicol. 16:1395–1404 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
