

Exploiting the mitochondrial unfolded protein response for cancer therapy in mice and human cells
- PMID: 21364280
- PMCID: PMC3069780
- DOI: 10.1172/JCI44855
Exploiting the mitochondrial unfolded protein response for cancer therapy in mice and human cells
Abstract
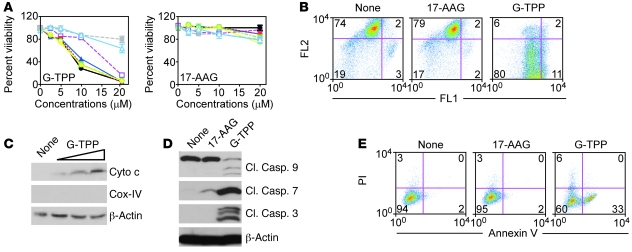
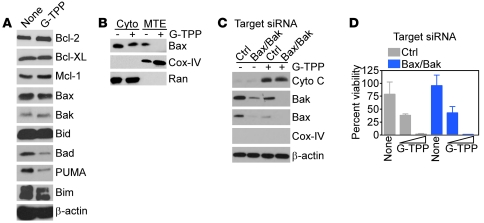
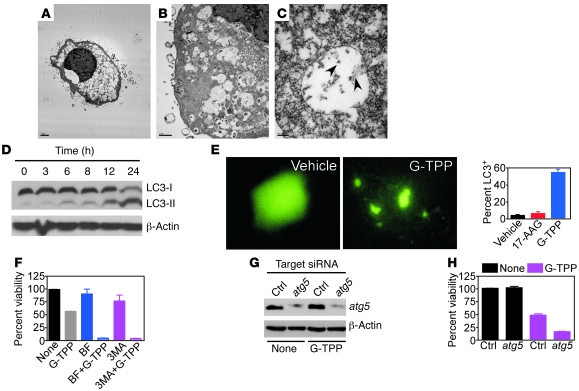
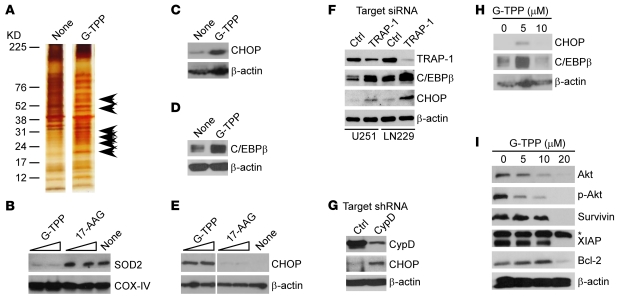
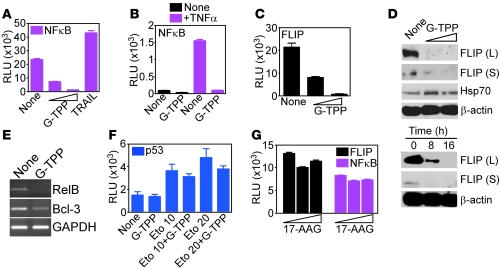
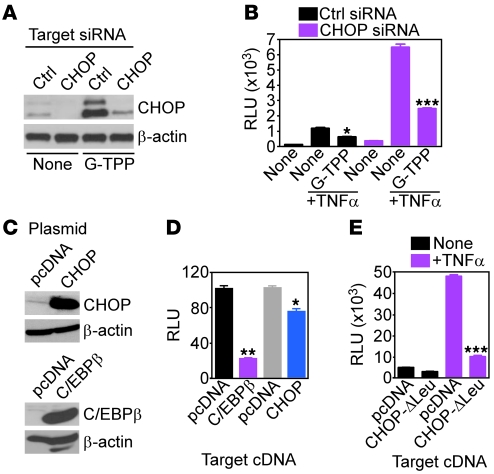
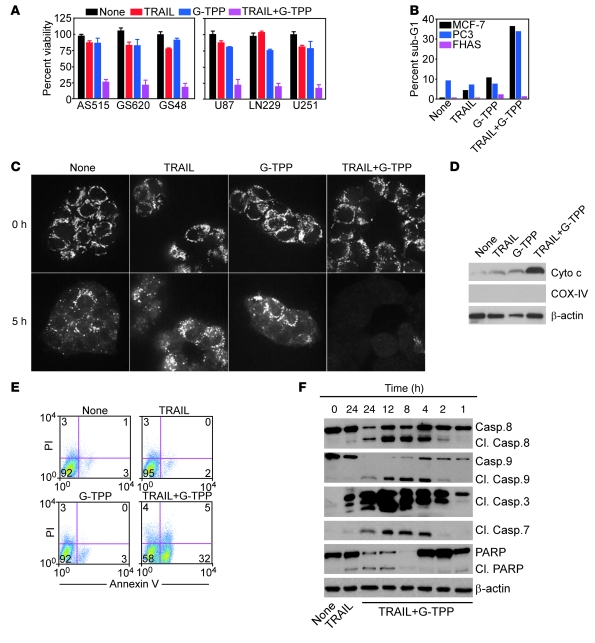
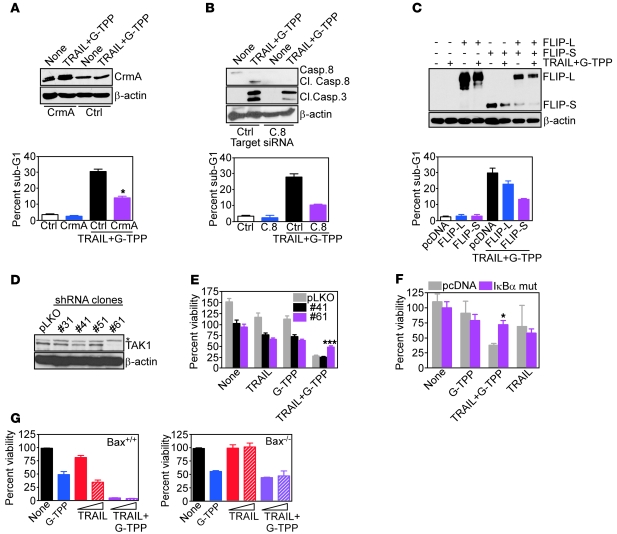
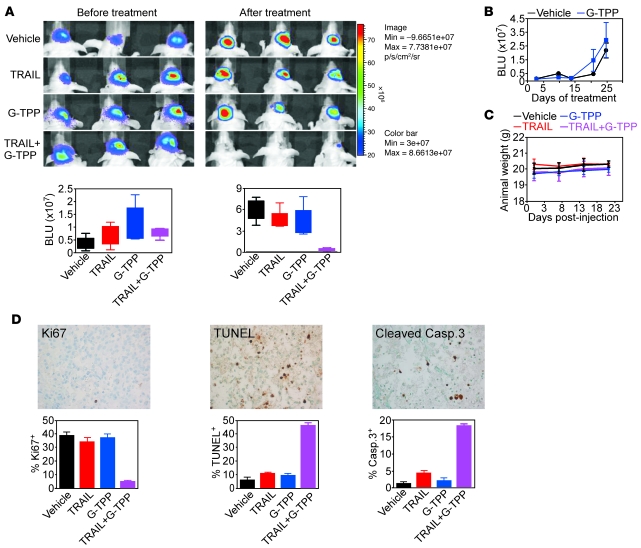
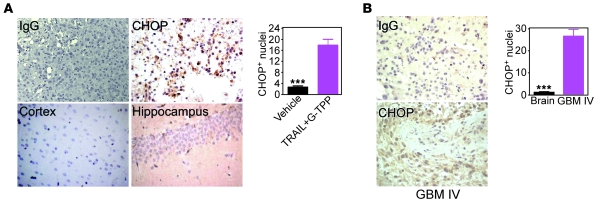
Fine tuning of the protein folding environment in subcellular organelles, such as mitochondria, is important for adaptive homeostasis and may participate in human diseases, but the regulators of this process are still largely elusive. Here, we have shown that selective targeting of heat shock protein-90 (Hsp90) chaperones in mitochondria of human tumor cells triggered compensatory autophagy and an organelle unfolded protein response (UPR) centered on upregulation of CCAAT enhancer binding protein (C/EBP) transcription factors. In turn, this transcriptional UPR repressed NF-κB-dependent gene expression, enhanced tumor cell apoptosis initiated by death receptor ligation, and inhibited intracranial glioblastoma growth in mice without detectable toxicity. These data reveal what we believe to be a novel role of Hsp90 chaperones in the regulation of the protein-folding environment in mitochondria of tumor cells. Disabling this general adaptive pathway could potentially be used in treatment of genetically heterogeneous human tumors.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
