

A Tbx1-Six1/Eya1-Fgf8 genetic pathway controls mammalian cardiovascular and craniofacial morphogenesis
- PMID: 21364285
- PMCID: PMC3069777
- DOI: 10.1172/JCI44630
A Tbx1-Six1/Eya1-Fgf8 genetic pathway controls mammalian cardiovascular and craniofacial morphogenesis
Erratum in
- J Clin Invest. 2011 May 2;121(5):2060
Abstract
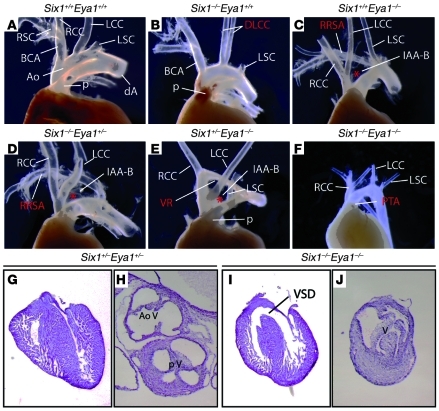
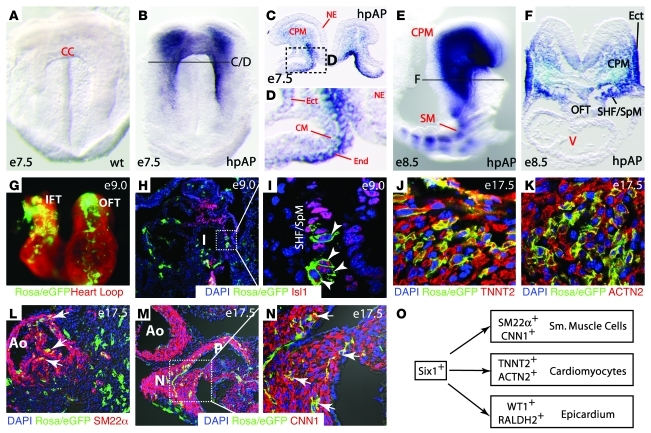
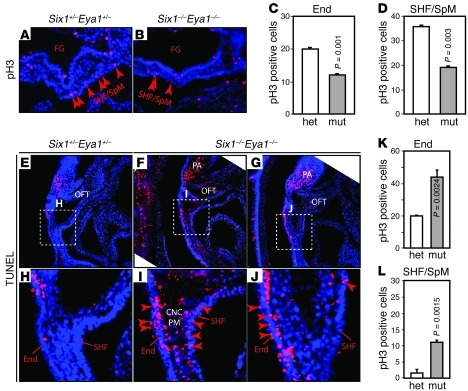
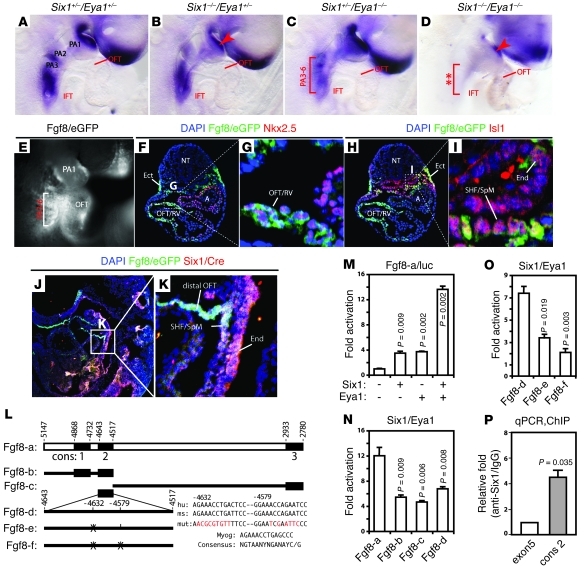
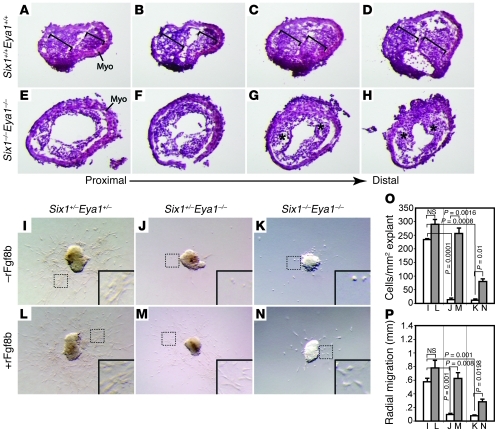
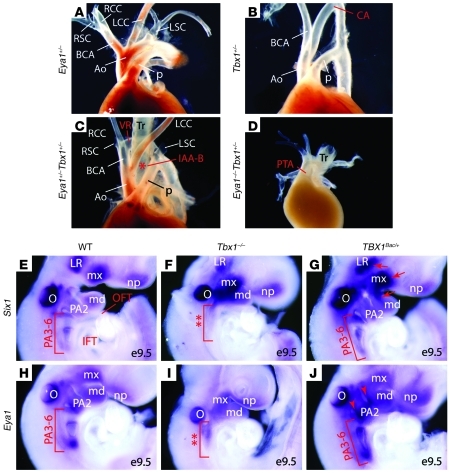
Shared molecular programs govern the formation of heart and head during mammalian embryogenesis. Development of both structures is disrupted in human chromosomal microdeletion of 22q11.2 (del22q11), which causes DiGeorge syndrome (DGS) and velo-cardio-facial syndrome (VCFS). Here, we have identified a genetic pathway involving the Six1/Eya1 transcription complex that regulates cardiovascular and craniofacial development. We demonstrate that murine mutation of both Six1 and Eya1 recapitulated most features of human del22q11 syndromes, including craniofacial, cardiac outflow tract, and aortic arch malformations. The mutant phenotypes were attributable in part to a reduction of fibroblast growth factor 8 (Fgf8), which was shown to be a direct downstream effector of Six1 and Eya1. Furthermore, we showed that Six1 and Eya1 genetically interacted with Fgf8 and the critical del22q11 gene T-box transcription factor 1 (Tbx1) in mice. Together, these findings reveal a Tbx1-Six1/Eya1-Fgf8 genetic pathway that is crucial for mammalian cardiocraniofacial morphogenesis and provide insights into the pathogenesis of human del22q11 syndromes.
Figures
References
-
- Waldo KL, et al. Conotruncal myocardium arises from a secondary heart field. Development. 2001;128(16):3179–3188. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
