

MEK-ERK-dependent multiple caspase activation by mitochondrial proapoptotic Bcl-2 family proteins is essential for heavy ion irradiation-induced glioma cell death
- PMID: 21364665
- PMCID: PMC3039836
- DOI: 10.1038/cddis.2010.37
MEK-ERK-dependent multiple caspase activation by mitochondrial proapoptotic Bcl-2 family proteins is essential for heavy ion irradiation-induced glioma cell death
Abstract
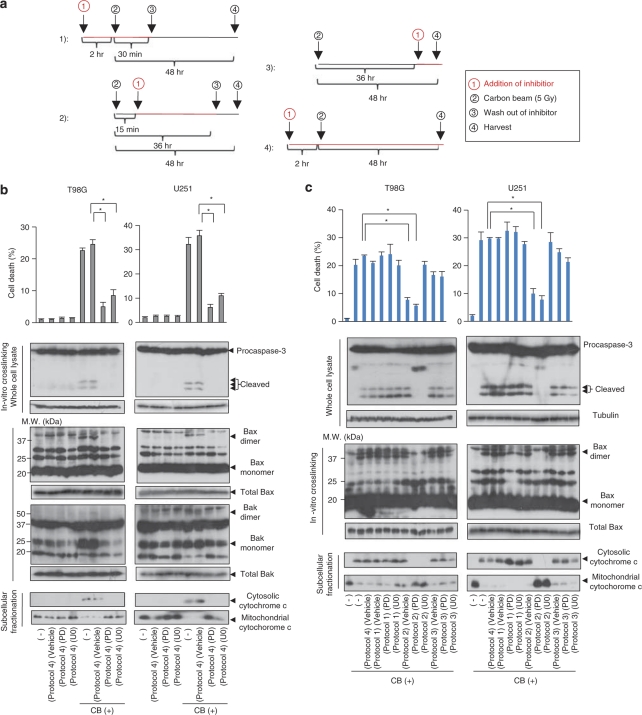
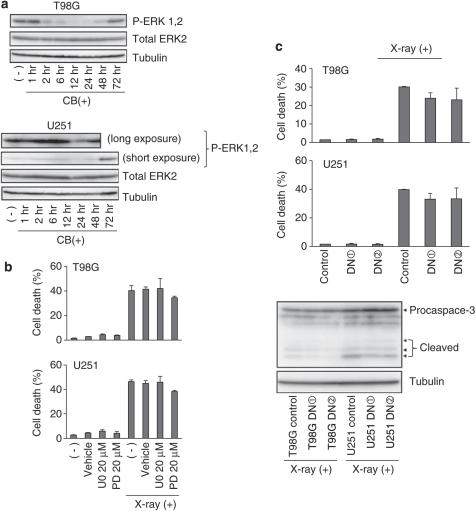
Recently developed heavy ion irradiation therapy using a carbon beam (CB) against systemic malignancy has numerous advantages. However, the clinical results of CB therapy against glioblastoma still have room for improvement. Therefore, we tried to clarify the molecular mechanism of CB-induced glioma cell death. T98G and U251 human glioblastoma cell lines were irradiated by CB, and caspase-dependent apoptosis was induced in both cell lines in a dose-dependent manner. Knockdown of Bax (BCL-2-associated X protein) and Bak (BCL-2-associated killer) and overexpression of Bcl-2 or Bcl-xl (B-cell lymphoma-extra large) showed the involvement of Bcl-2 family proteins upstream of caspase activation, including caspase-8, in CB-induced glioma cell death. We also detected the activation of extracellular signal-regulated kinase (ERK) and the knockdown of ERK regulator mitogen-activated protein kinase kinase (MEK)1/2 or overexpression of a dominant-negative (DN) ERK inhibited CB-induced glioma cell death upstream of the mitochondria. In addition, application of MEK-specific inhibitors for defined periods showed that the recovery of activation of ERK between 2 and 36 h after irradiation is essential for CB-induced glioma cell death. Furthermore, MEK inhibitors or overexpression of a DN ERK failed to significantly inhibit X-ray-induced T98G and U251 cell death. These results suggested that the MEK-ERK cascade has a crucial role in CB-induced glioma cell death, which is known to have a limited contribution to X-ray-induced glioma cell death.
Figures





References
-
- Fukumura A, Tsujii H, Kamada T, Baba M, Tsuji H, Kato H, et al. Carbon-ion radiotherapy: clinical aspects and related dosimetry. Radiat Prot Dosimetry. 2009;137:149–155. - PubMed
-
- Tsujii H, Mizoe JE, Kamada T, Baba M, Kato S, Kato H, et al. Overview of clinical experiences on carbon ion radiotherapy at NIRS. Radiother Oncol. 2004;73 (Suppl 2:S41–S49. - PubMed
-
- Blakely EA, Kronenberg A. Heavy-ion radiobiology: new approaches to delineate mechanisms underlying enhanced biological effectiveness. Radiat Res. 1998;150 (5 Suppl:S126–S145. - PubMed
-
- Masunaga S, Ando K, Uzawa A, Hirayama R, Furusawa Y, Koike S, et al. Radiobiologic significance of response of intratumor quiescent cells in vivo to accelerated carbon ion beams compared with gamma-rays and reactor neutron beams. Int J Radiat Oncol Biol Phys. 2008;70:221–228. - PubMed
-
- Suzuki M, Kase Y, Yamaguchi H, Kanai T, Ando K. Relative biological effectiveness for cell-killing effect on various human cell lines irradiated with heavy-ion medical accelerator in Chiba (HIMAC) carbon-ion beams. Int J Radiat Oncol Biol Phys. 2000;48:241–250. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
