

Using genomic sequencing for classical genetics in E. coli K12
- PMID: 21364914
- PMCID: PMC3045373
- DOI: 10.1371/journal.pone.0016717
Using genomic sequencing for classical genetics in E. coli K12
Abstract
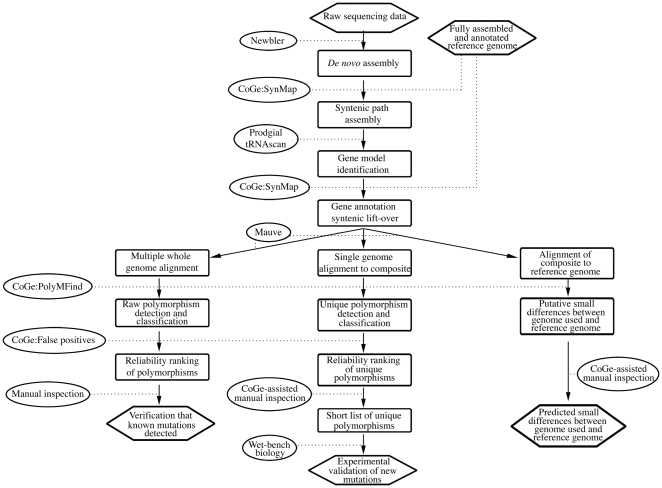
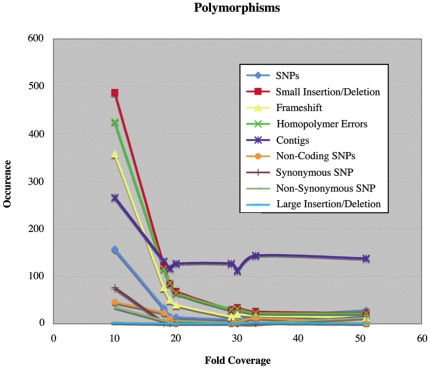
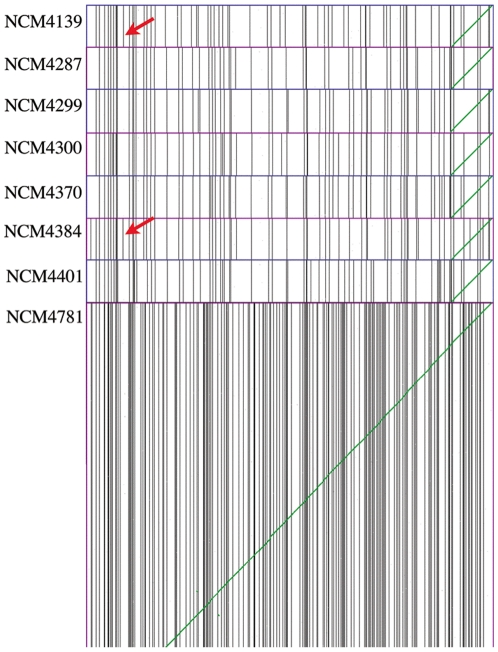
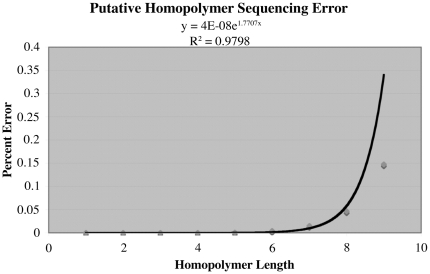
We here develop computational methods to facilitate use of 454 whole genome shotgun sequencing to identify mutations in Escherichia coli K12. We had Roche sequence eight related strains derived as spontaneous mutants in a background without a whole genome sequence. They provided difference tables based on assembling each genome to reference strain E. coli MG1655 (NC_000913). Due to the evolutionary distance to MG1655, these contained a large number of both false negatives and positives. By manual analysis of the dataset, we detected all the known mutations (24 at nine locations) and identified and genetically confirmed new mutations necessary and sufficient for the phenotypes we had selected in four strains. We then had Roche assemble contigs de novo, which we further assembled to full-length pseudomolecules based on synteny with MG1655. This hybrid method facilitated detection of insertion mutations and allowed annotation from MG1655. After removing one genome with less than the optimal 20- to 30-fold sequence coverage, we identified 544 putative polymorphisms that included all of the known and selected mutations apart from insertions. Finally, we detected seven new mutations in a total of only 41 candidates by comparing single genomes to composite data for the remaining six and using a ranking system to penalize homopolymer sequencing and misassembly errors. An additional benefit of the analysis is a table of differences between MG1655 and a physiologically robust E. coli wild-type strain NCM3722. Both projects were greatly facilitated by use of comparative genomics tools in the CoGe software package (http://genomevolution.org/).
Conflict of interest statement
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
