

Neisseria meningitidis is structured in clades associated with restriction modification systems that modulate homologous recombination
- PMID: 21368196
- PMCID: PMC3060241
- DOI: 10.1073/pnas.1019751108
Neisseria meningitidis is structured in clades associated with restriction modification systems that modulate homologous recombination
Abstract
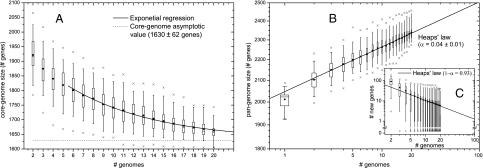
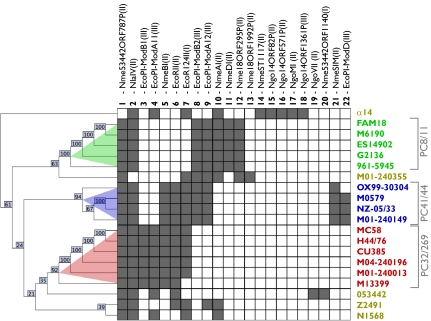
Molecular data on a limited number of chromosomal loci have shown that the population of Neisseria meningitidis (Nm), a deadly human pathogen, is structured in distinct lineages. Given that the Nm population undergoes substantial recombination, the mechanisms resulting in the evolution of these lineages, their persistence in time, and the implications for the pathogenicity of the bacterium are not yet completely understood. Based on whole-genome sequencing, we show that Nm is structured in phylogenetic clades. Through acquisition of specific genes and through insertions and rearrangements, each clade has acquired and remodeled specific genomic tracts, with the potential to impact on the commensal and virulence behavior of Nm. Despite this clear evidence of a structured population, we confirm high rates of detectable recombination throughout the whole Nm chromosome. However, gene conversion events were found to be longer within clades than between clades, suggesting a DNA cleavage mechanism associated with the phylogeny of the species. We identify 22 restriction modification systems, probably acquired by horizontal gene transfer from outside of the species/genus, whose distribution in the different strains coincides with the phylogenetic clade structure. We provide evidence that these clade-associated restriction modification systems generate a differential barrier to DNA exchange consistent with the observed population structure. These findings have general implications for the emergence of lineage structure and virulence in recombining bacterial populations, and they could provide an evolutionary framework for the population biology of a number of other bacterial species that show contradictory population structure and dynamics.
Conflict of interest statement
Conflict of interest statement: M.C., A.C., M.P., R.R., D.S., K.L.S., and D.M. are employees of Novartis Vaccines; S.B. and E.S. have PhD grants from Novartis Vaccines; and E.R.M. serves on the Scientific Advisory Board of Novartis Vaccines.
Figures



References
-
- Stephens DS, Greenwood B, Brandtzaeg P. Epidemic meningitis, meningococcaemia, and Neisseria meningitidis. Lancet. 2007;369:2196–2210. - PubMed
-
- Maiden MC. Population structure of Neisseria meningitidis. In: Ferreirós C, Criado MT, Vázquez J, editors. Emerging Strategies in the Fight Against Meningitis: Molecular and Cellular Aspects. Wymondham, Norfolk, United Kingdom: Horizon Scientific Press; 2002. pp. 151–170.
-
- Dunning Hotopp JC, et al. Comparative genomics of Neisseria meningitidis: Core genome, islands of horizontal transfer and pathogen-specific genes. Microbiology. 2006;152:3733–3749. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
