

Bortezomib interacts synergistically with belinostat in human acute myeloid leukaemia and acute lymphoblastic leukaemia cells in association with perturbations in NF-κB and Bim
- PMID: 21375523
- PMCID: PMC3358826
- DOI: 10.1111/j.1365-2141.2011.08591.x
Bortezomib interacts synergistically with belinostat in human acute myeloid leukaemia and acute lymphoblastic leukaemia cells in association with perturbations in NF-κB and Bim
Abstract
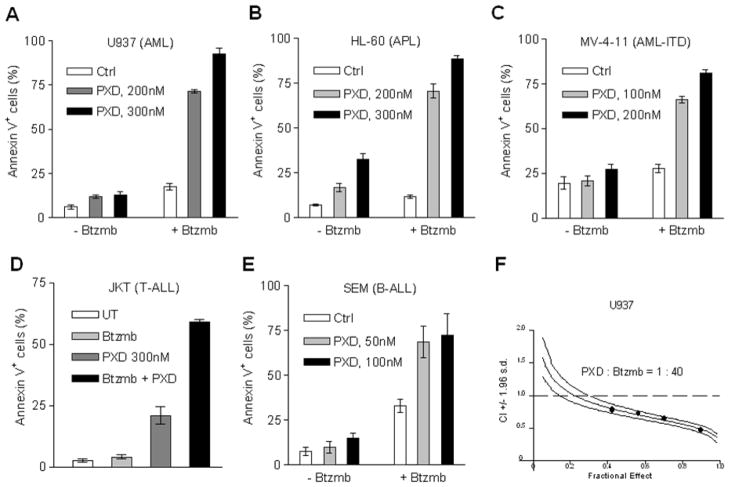
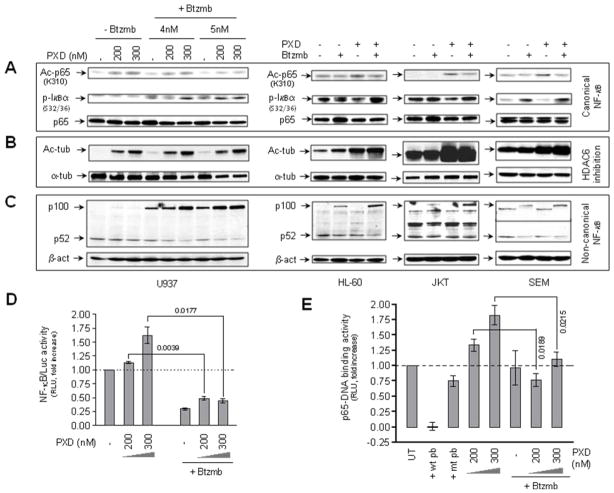
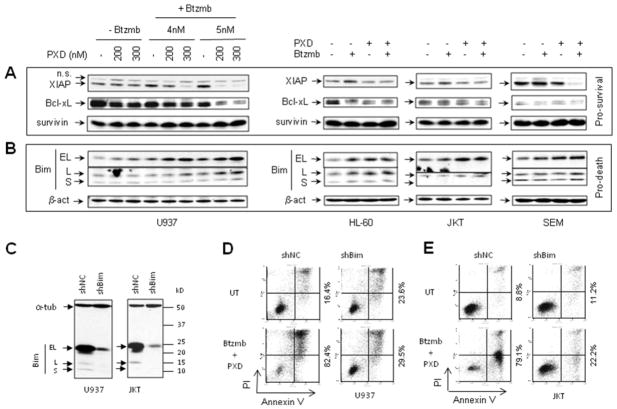
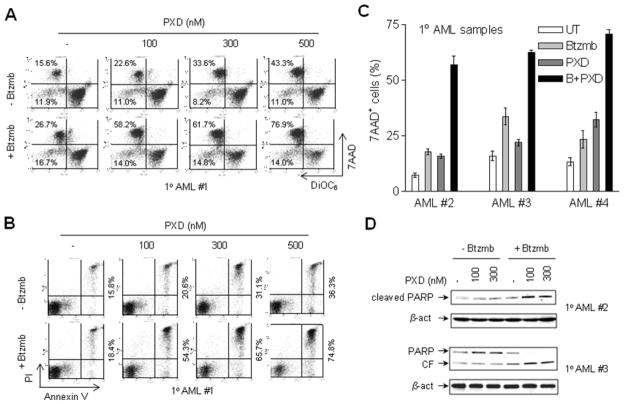
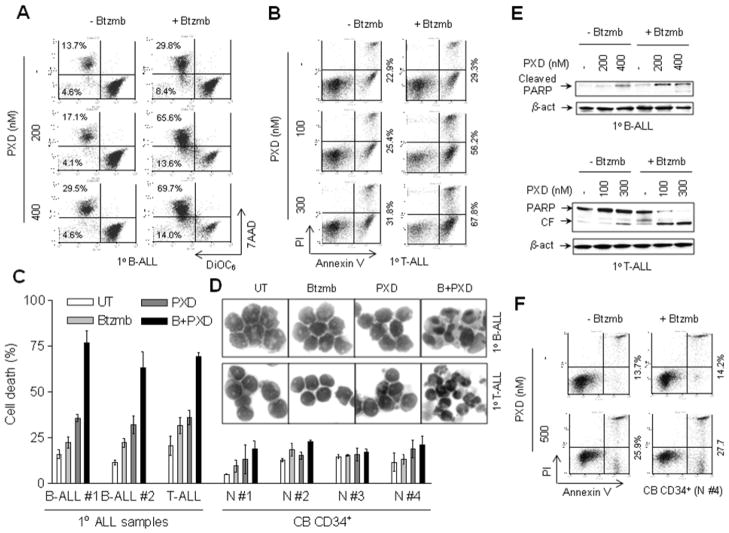
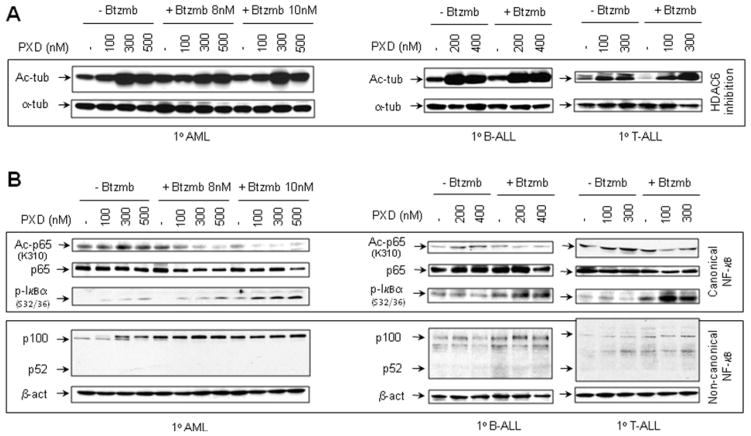
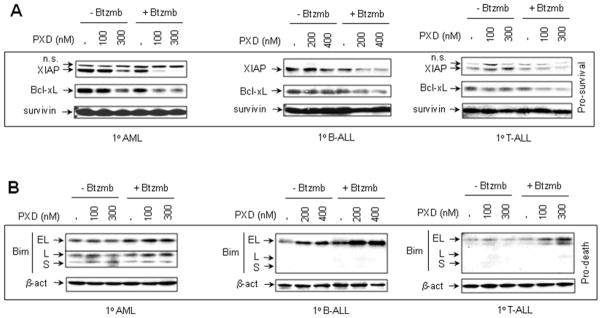
Interactions between the histone deacetylase inhibitor belinostat and the proteasome inhibitor bortezomib were investigated in acute myeloid leukaemia (AML) and acute lymphoblastic leukaemia (ALL) cells. Co-administration of sub-micromolar concentrations of belinostat with low nanomolar concentrations of bortezomib sharply increased apoptosis in both AML and ALL cell lines and primary blasts. Synergistic interactions were associated with interruption of both canonical and non-canonical nuclear factor (NF)-κB signalling pathways, e.g. accumulation of the phosphorylated (S32/S36) form of IκBα, diminished belinostat-mediated RelA/p65 hyperacetylation (K310), and reduced processing of p100 into p52. These events were accompanied by down-regulation of NF-κB-dependent pro-survival proteins (e.g. XIAP, Bcl-xL). Moreover, belinostat/bortezomib co-exposure induced up-regulation of the BH3-only pro-death protein Bim. Significantly, shRNA knock-down of Bim substantially reduced the lethality of belinostat/bortezomib regimens. Administration of belinostat ± bortezomib also induced hyperacetylation (K40) of α-tubulin, indicating histone deacetylase inhibitor 6 inhibition. Finally, in contrast to the pronounced lethality of belinostat/bortezomib toward primary leukaemia blasts, equivalent treatment was relatively non-toxic to normal CD34(+) cells. Together, these findings indicate that belinostat and bortezomib interact synergistically in both cultured and primary AML and ALL cells, and raise the possibilities that up-regulation of Bim and interference with NF-κB pathways contribute to this phenomenon. They also suggest that combined belinostat/bortezomib regimens warrant further attention in acute leukaemias.
© 2011 Blackwell Publishing Ltd.
Conflict of interest statement
Figures
References
-
- Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5(9):769–784. - PubMed
-
- Drummond DC, Noble CO, Kirpotin DB, Guo Z, Scott GK, Benz CC. Clinical development of histone deacetylase inhibitors as anticancer agents. Annu Rev Pharmacol Toxicol. 2005;45:495–528. - PubMed
-
- Grant S, Easley C, Kirkpatrick P. Vorinostat. Nature Review Drug Discovery. 2007;6:1–2. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
