

Review
doi: 10.1161/CIRCULATIONAHA.109.914838.
Genetic testing for potentially lethal, highly treatable inherited cardiomyopathies/channelopathies in clinical practice
Affiliations
- PMID: 21382904
- PMCID: PMC3073829
- DOI: 10.1161/CIRCULATIONAHA.109.914838
Item in Clipboard
Review
Genetic testing for potentially lethal, highly treatable inherited cardiomyopathies/channelopathies in clinical practice
Circulation.
.
No abstract available
Conflict of interest statement
Figures
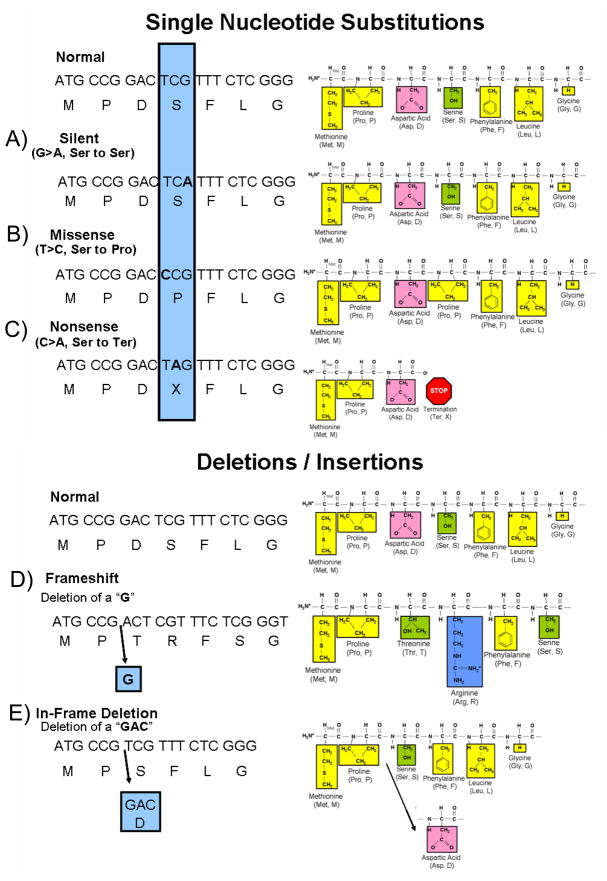
Compared to the depicted normal DNA, amino acid (single letter abbreviation) sequence and resulting peptide sequence are examples of nucleotide substitutions and deletion mutations. The amino acids of the peptide sequence are color coded to represent their unique biophysical properties where yellow represents nonpolar hydrophobic amino acids, green are polar hydrophilic, pink are negatively charged acidic residues, and blue represents positively charged basic amino acid residues. A nucleotide change resulting in a new codon that encodes for: A) the same amino acid as the normal sequence is a silent mutation, B) a different amino acid is a missense mutation, and C) for a termination codon is a nonsense mutation. Illustrated in D) is a deletion of a single nucleotide (G) that results in a shift of the open reading frame of the transcript thus representing a frameshift mutation. Note how the sequence of amino acids has been altered from this point forward. Although not illustrated here, frameshift mutations as a result of a deletion or insertion of nucleotides often lead to a premature stop codon and thus a truncated protein. Illustrated in E), the deletion of three nucleotides (GAC) produced an in-frame deletion of a single amino acid (aspartic acid, Asp) in the protein. The remaining amino acid sequence is unaltered. Three nucleotide insertions (not shown) can have a similar affect whereby an amino acid is inserted into the protein product. Figure adapted from Tester and Ackerman
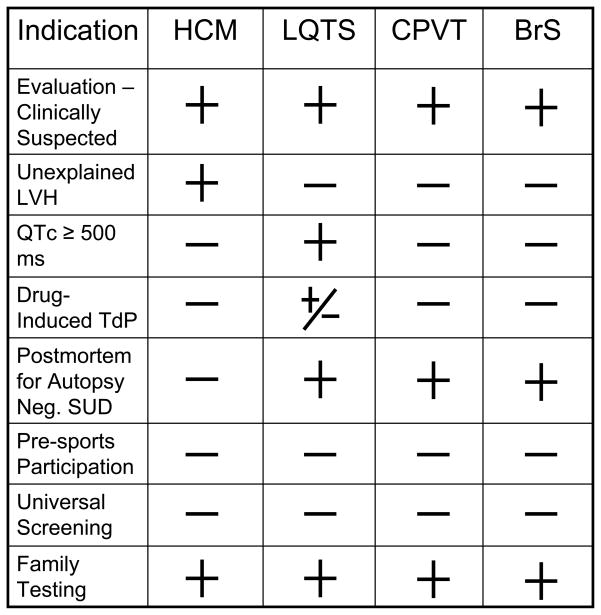
Provided is a table of possible indications for genetic testing for hypertrophic cardiomyopathy (HCM), long QT syndrome (LQTS), catecholaminergic polymorphic ventricular tachycardia (CPVT), and Brugada syndrome (BrS). LVH = left ventricular hypertrophy, TdP = torsade de pointes, SUD = sudden unexplained death. The + symbol represents a positive indication for genetic testing. The − symbol represents an indicator that does not warrant for genetic testing for the specific disorder. The +/− symbol represents an indicator that may or may not warrant genetic testing.
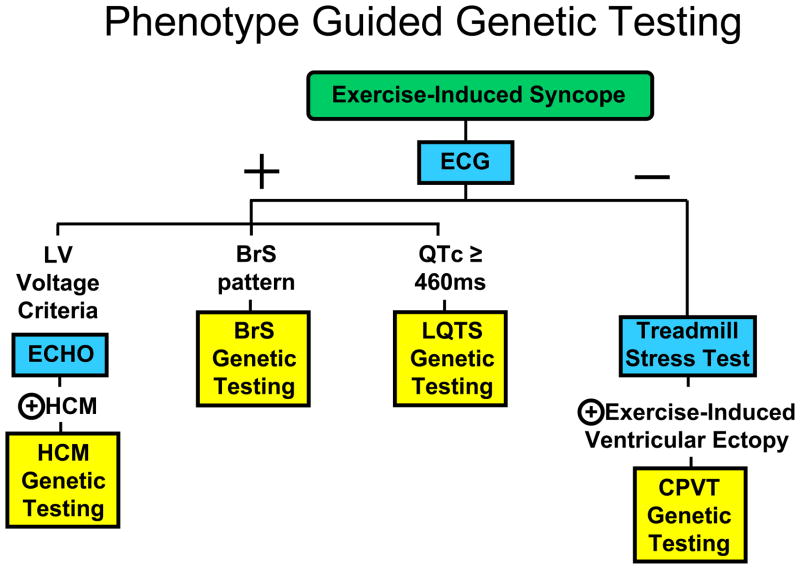
Depicted for illustrative purposes is a work flow decision tree for indicating which genetic test would be appropriate to order based on the clinical evaluation of an individual manifesting with exercise-induced syncope. The blue boxes represent specific clinical evaluation tests. The yellow boxes represent specific genetic testing panels. The + symbol represents a positive evaluation and the − symbol represents a negative evaluation for the respective clinical test. The key point is that genetic testing for these disorders should be phenotype-guided, NOT universal.

Shown is the current diagnostic, prognostic, and therapeutic utility of genetic testing for hypertrophic cardiomyopathy (HCM), long QT syndrome (LQTS), catecholaminergic polymorphic ventricular tachycardia (CPVT), and Brugada syndrome (BrS). The + symbol indicates the test has utility, the − symbol indicates no current measurable utility, and +/− indicates the test may have some utility.
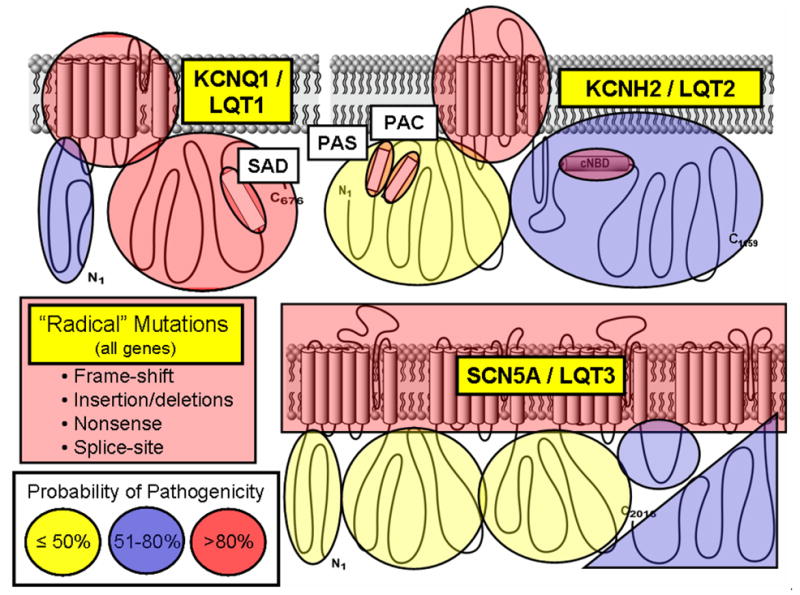
Depicted are the three major ion channels causative for LQTS with areas of probability of pathogenicity shown for mutations localizing to these respective areas. While “radical” mutations have a greater then 90% probability of being a true pathogenic mutation, the level of probability for missense mutations vary depending on their location for each channel protein. Missense mutations residing in red shaded areas have a high probability (>80%) of being pathogenic, those in blue are possibly (51–80%) pathogenic, and those in yellow shaded areas truly represent variants of uncertain significance (VUS, ≤50% probability) clinically. Figure adapted from Tester and Ackerman.
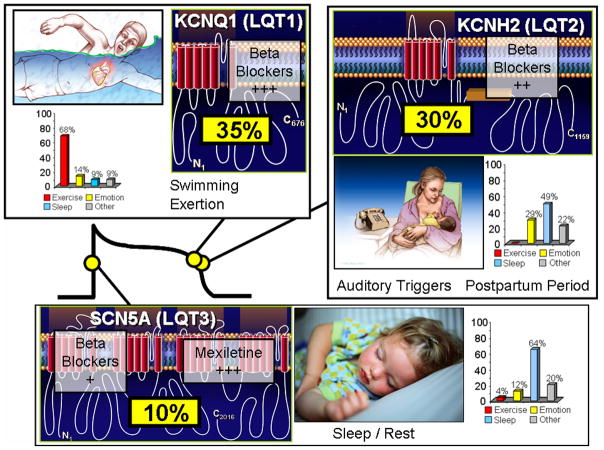
Seventy-five percent of clinically strong LQTS is due to mutations in three genes (35% KCNQ1, 30% KCNH2, and 10% in SCN5A) encoding for ion channels that are critically responsible for the orchestration of the cardiac action potential. Genotype-phenotype correlations have been observed, including swimming/exertion and LQT1, auditory triggers/postpartum period and LQT2, and sleep/rest and LQT3. The bar graphs represent genotype-phenotype data from reference . Also illustrated is the relative gene-specific effectiveness in β blocker therapy where β blockers are extremely protective in LQT1 patients, moderately protective in LQT2, and may not provide sufficient protection for those with LQT3. The late sodium current blockers like mexiletine, ranolazine, and propranolol may be protective in LQT3.
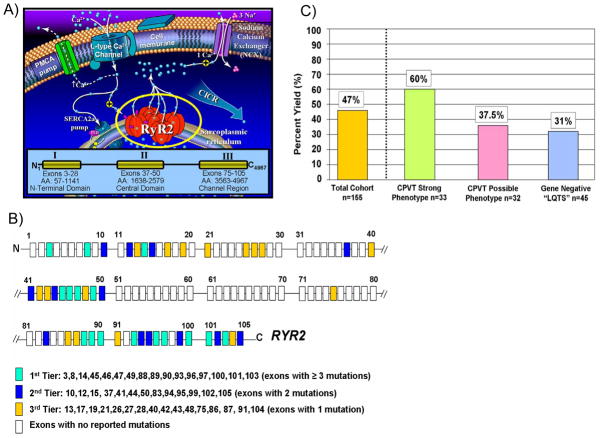
A) Perturbations in key components of the calcium-induced calcium release (CICR) mechanism responsible for cardiac excitation-contraction coupling are the pathogenic basis for CPVT. At the center of this mechanism is the RYR2-encoded cardiac ryanodine receptor/calcium release channel located in sarcoplasmic reticulum membrane. Mutations in RyR2 are clustered and distributed in three “hot-spot” regions of this 4967 amino acid (AA) protein; Domain I or N-terminal Domain (AA 57-1141), Domain II or the Central Domain (AA 1638-2579), and Domain III or Channel Region (AA 3563-4967). B) Because of this clustering of mutations, a tiered strategy for genetic testing of the 105 exon RYR2 gene has been proposed. C) Illustrated is the percent yield of RYR2 mutation detection in cases of strong CPVT, possible CPVT, and LQT1-3 gene-negative “LQTS” referral cases manifesting with exercise-induced cardiac events.
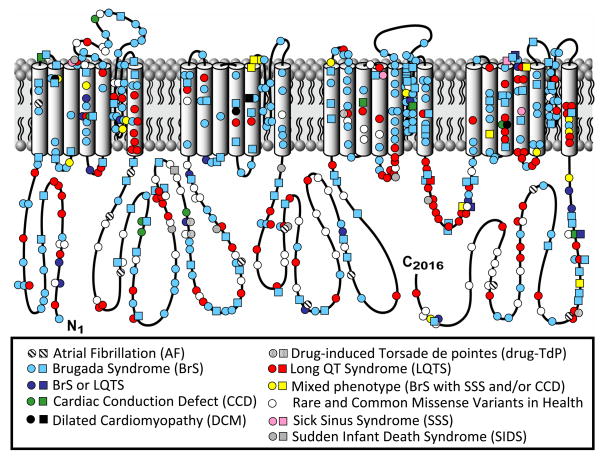
Depicted is the linear topology of the 2016-amino acid containing cardiac sodium channel isoform with mutation location and their associated disorders. Circles denote missense mutations and squares represent “radical” mutations including, frame-shift insertion/deletion mutations, in-frame insertions/deletions, nonsense, and splice-site mutations. Figure adapted from Tester and Ackerman.
Similar articles
-
Personalized medicine: genetic diagnosis for inherited cardiomyopathies/channelopathies.Rev Esp Cardiol (Engl Ed). 2013 Apr;66(4):298-307. doi: 10.1016/j.rec.2012.12.010. Epub 2013 Feb 26. Rev Esp Cardiol (Engl Ed). 2013. PMID: 24775620 Review.
-
Genetic basis of channelopathies and cardiomyopathies in Hong Kong Chinese patients: a 10-year regional laboratory experience.Hong Kong Med J. 2018 Aug;24(4):340-349. doi: 10.12809/hkmj176870. Epub 2018 Mar 2. Hong Kong Med J. 2018. PMID: 29497013
-
HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA).Heart Rhythm. 2011 Aug;8(8):1308-39. doi: 10.1016/j.hrthm.2011.05.020. Heart Rhythm. 2011. PMID: 21787999 No abstract available.
-
HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA).Europace. 2011 Aug;13(8):1077-109. doi: 10.1093/europace/eur245. Europace. 2011. PMID: 21810866 No abstract available.
-
Establishment of Specialized Clinical Cardiovascular Genetics Programs: Recognizing the Need and Meeting Standards: A Scientific Statement From the American Heart Association.Circ Genom Precis Med. 2019 Jun;12(6):e000054. doi: 10.1161/HCG.0000000000000054. Epub 2019 May 23. Circ Genom Precis Med. 2019. PMID: 31117808
Cited by
-
Heart failure: advanced development in genetics and epigenetics.Biomed Res Int. 2015;2015:352734. doi: 10.1155/2015/352734. Epub 2015 Apr 9. Biomed Res Int. 2015. PMID: 25949994 Free PMC article. Review.
-
Genetic testing in heritable cardiac arrhythmia syndromes: differentiating pathogenic mutations from background genetic noise.Curr Opin Cardiol. 2013 Jan;28(1):63-71. doi: 10.1097/HCO.0b013e32835b0a41. Curr Opin Cardiol. 2013. PMID: 23128497 Free PMC article. Review.
-
Almanac 2011: Cardiac Arrhythmias and Pacing. The National Society Journals Present Selected Research that has Driven Recent Advances in Clinical Cardiology.Mater Sociomed. 2011;23(3):147-60. doi: 10.5455/msm.2011.23.147-160. Mater Sociomed. 2011. PMID: 23922508 Free PMC article. No abstract available.
-
HRS policy statement: clinical cardiac electrophysiology fellowship curriculum: update 2011.Heart Rhythm. 2011 Aug;8(8):1340-56. doi: 10.1016/j.hrthm.2011.06.008. Epub 2011 Jun 14. Heart Rhythm. 2011. PMID: 21699868 Free PMC article. No abstract available.
-
Genetics of hypertrophic cardiomyopathy: advances and pitfalls in molecular diagnosis and therapy.Appl Clin Genet. 2014 Oct 3;7:195-208. doi: 10.2147/TACG.S49126. eCollection 2014. Appl Clin Genet. 2014. PMID: 25328416 Free PMC article. Review.
References
-
- Nussbaum RLMR, Willard HF. Thompson & Thompson Genetics in Medicine. 6. Philadelphia, Pa: WB Saunders Co; 2001. pp. 51–94.
-
- den Dunnen JT, Antonarakis SE. Nomenclature for the description of human sequence variations. Human Genetics. 2001;109(1):121–124. - PubMed
-
- Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2(5):507–517. - PubMed
-
- Tester DJ, Ackerman MJ. Genetic Testing. In: Gussak I, Antzelevitch C, editors. Electrical diseases of the heart: genetics, mechanisms, treatment, prevention. London: Springer; 2008. pp. 444–458.
-
- Miller TE, Estrella E, Myerburg RJ, Garcia de Viera J, Moreno N, Rusconi P, Ahearn ME, Baumbach L, Kurlansky P, Wolff G, Bishopric NH. Recurrent third-trimester fetal loss and maternal mosaicism for long-QT syndrome.[see comment] Circulation. 2004;109(24):3029–3034. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
