

High resolution mapping of Twist to DNA in Drosophila embryos: Efficient functional analysis and evolutionary conservation
- PMID: 21383317
- PMCID: PMC3065704
- DOI: 10.1101/gr.104018.109
High resolution mapping of Twist to DNA in Drosophila embryos: Efficient functional analysis and evolutionary conservation
Abstract
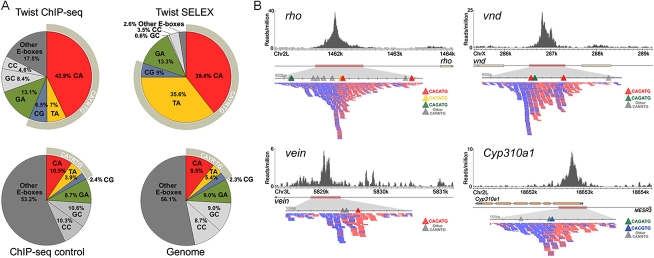
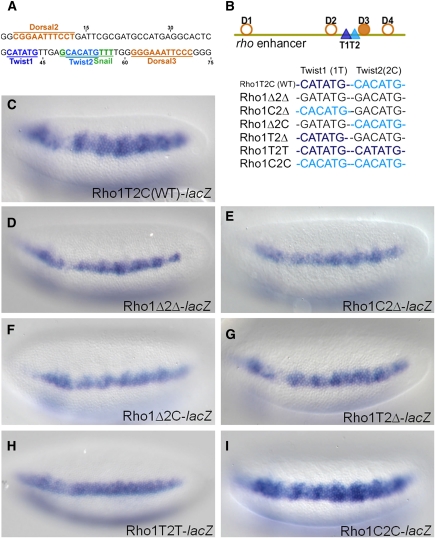
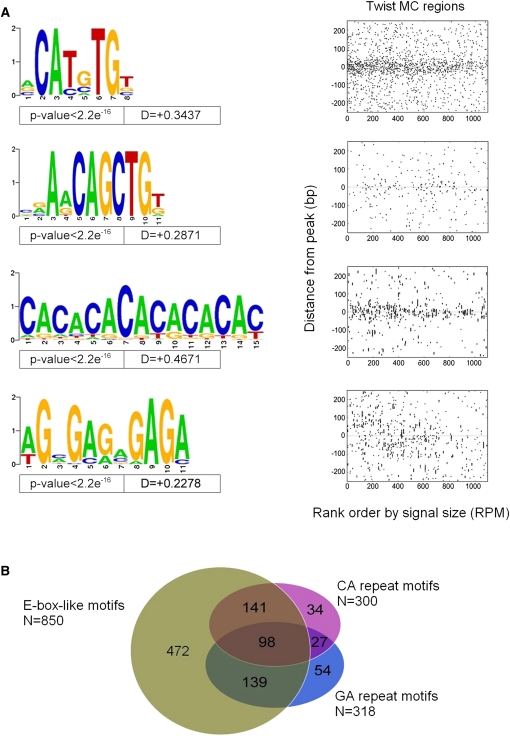
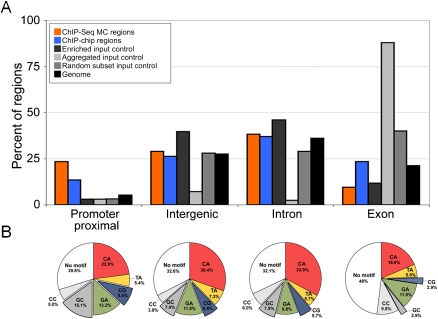
Cis-regulatory modules (CRMs) function by binding sequence specific transcription factors, but the relationship between in vivo physical binding and the regulatory capacity of factor-bound DNA elements remains uncertain. We investigate this relationship for the well-studied Twist factor in Drosophila melanogaster embryos by analyzing genome-wide factor occupancy and testing the functional significance of Twist occupied regions and motifs within regions. Twist ChIP-seq data efficiently identified previously studied Twist-dependent CRMs and robustly predicted new CRM activity in transgenesis, with newly identified Twist-occupied regions supporting diverse spatiotemporal patterns (>74% positive, n = 31). Some, but not all, candidate CRMs require Twist for proper expression in the embryo. The Twist motifs most favored in genome ChIP data (in vivo) differed from those most favored by Systematic Evolution of Ligands by EXponential enrichment (SELEX) (in vitro). Furthermore, the majority of ChIP-seq signals could be parsimoniously explained by a CABVTG motif located within 50 bp of the ChIP summit and, of these, CACATG was most prevalent. Mutagenesis experiments demonstrated that different Twist E-box motif types are not fully interchangeable, suggesting that the ChIP-derived consensus (CABVTG) includes sites having distinct regulatory outputs. Further analysis of position, frequency of occurrence, and sequence conservation revealed significant enrichment and conservation of CABVTG E-box motifs near Twist ChIP-seq signal summits, preferential conservation of ±150 bp surrounding Twist occupied summits, and enrichment of GA- and CA-repeat sequences near Twist occupied summits. Our results show that high resolution in vivo occupancy data can be used to drive efficient discovery and dissection of global and local cis-regulatory logic.
Figures



References
-
- Arnosti DN, Kulkarni MM 2005. Transcriptional enhancers: Intelligent enhanceosomes or flexible billboards? J Cell Biochem 94: 890–898 - PubMed
-
- Bergman CM, Pfeiffer BD, Rincon-Limas DE, Hoskins RA, Gnirke A, Mungall CJ, Wang AM, Kronmiller B, Pacleb J, Park S, et al. 2002. Assessing the impact of comparative genomic sequence data on the functional annotation of the Drosophila genome. Genome Biol 3: RESEARCH0086 doi: 10.1186/gb-2002-3-12-research0086 - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases