

Comparing genotyping algorithms for Illumina's Infinium whole-genome SNP BeadChips
- PMID: 21385424
- PMCID: PMC3063825
- DOI: 10.1186/1471-2105-12-68
Comparing genotyping algorithms for Illumina's Infinium whole-genome SNP BeadChips
Abstract
Background: Illumina's Infinium SNP BeadChips are extensively used in both small and large-scale genetic studies. A fundamental step in any analysis is the processing of raw allele A and allele B intensities from each SNP into genotype calls (AA, AB, BB). Various algorithms which make use of different statistical models are available for this task. We compare four methods (GenCall, Illuminus, GenoSNP and CRLMM) on data where the true genotypes are known in advance and data from a recently published genome-wide association study.
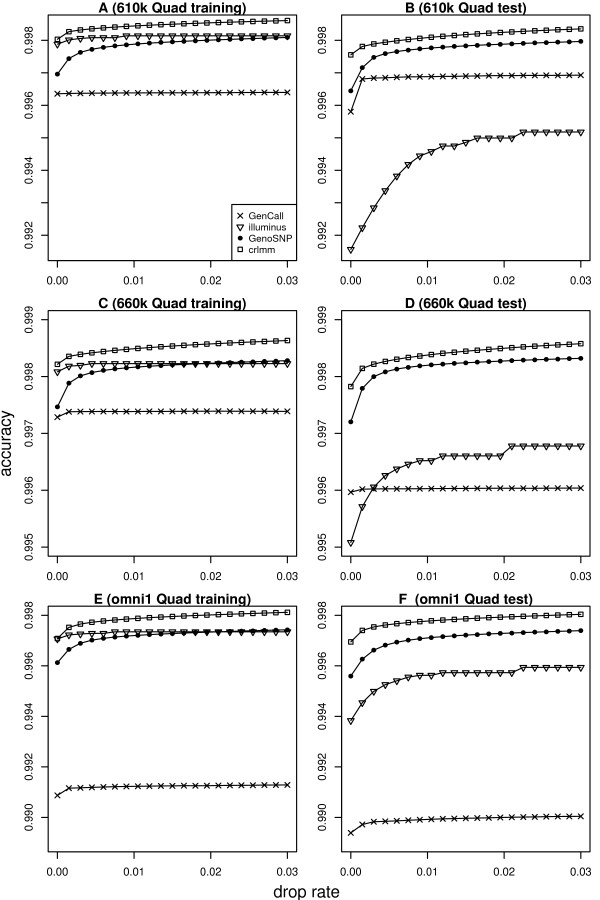
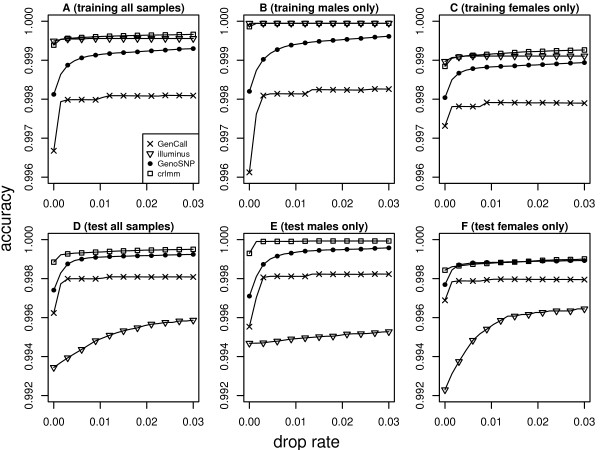
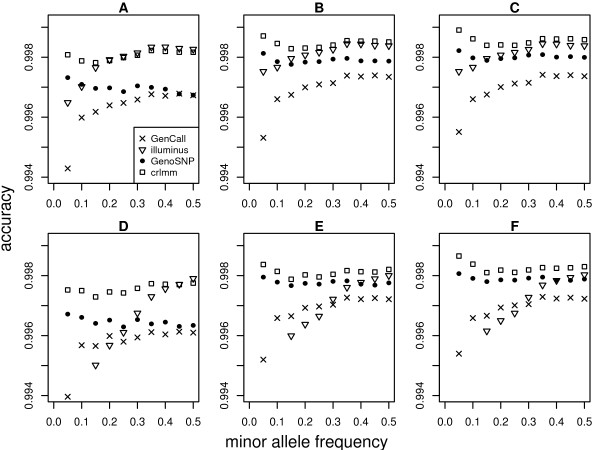
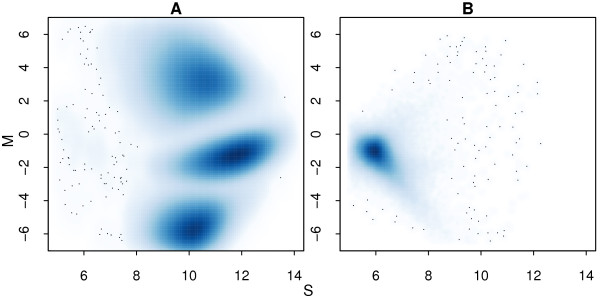
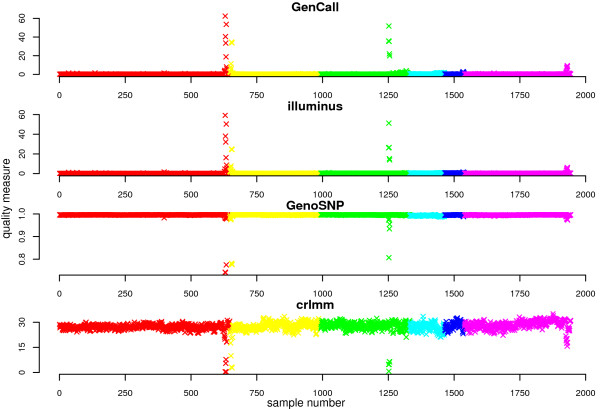
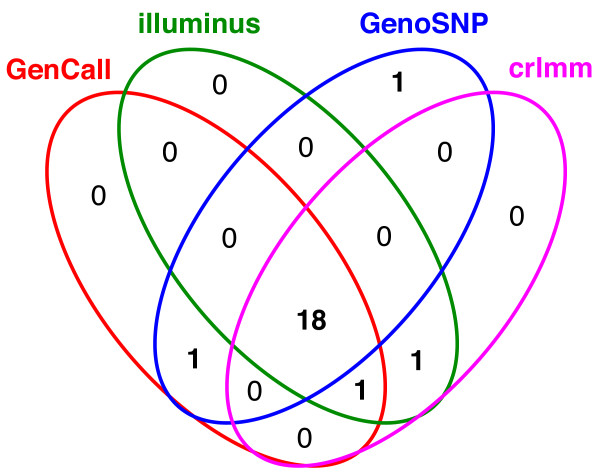
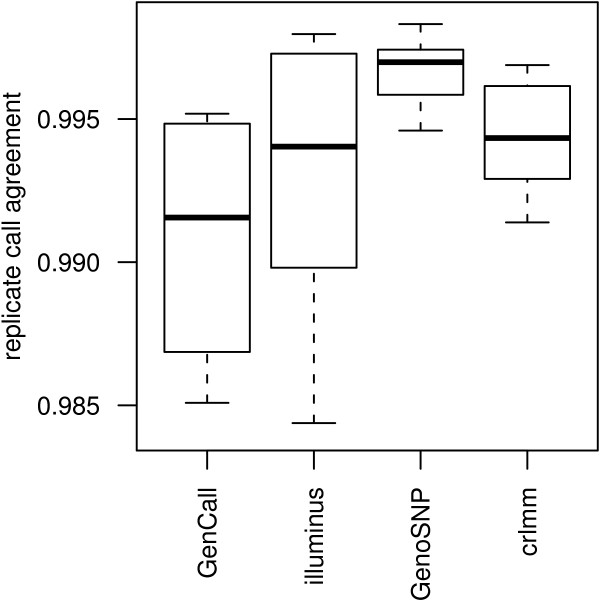
Results: In general, differences in accuracy are relatively small between the methods evaluated, although CRLMM and GenoSNP were found to consistently outperform GenCall. The performance of Illuminus is heavily dependent on sample size, with lower no call rates and improved accuracy as the number of samples available increases. For X chromosome SNPs, methods with sex-dependent models (Illuminus, CRLMM) perform better than methods which ignore gender information (GenCall, GenoSNP). We observe that CRLMM and GenoSNP are more accurate at calling SNPs with low minor allele frequency than GenCall or Illuminus. The sample quality metrics from each of the four methods were found to have a high level of agreement at flagging samples with unusual signal characteristics.
Conclusions: CRLMM, GenoSNP and GenCall can be applied with confidence in studies of any size, as their performance was shown to be invariant to the number of samples available. Illuminus on the other hand requires a larger number of samples to achieve comparable levels of accuracy and its use in smaller studies (50 or fewer individuals) is not recommended.
Figures

References
-
- Peiffer D, Le J, Steemers F, Chang W, Jenniges T, Garcia F, Haden K, Li J, Shaw C, Belmont J, Cheung S, Shen R, Barker D, Gunderson K. High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res. 2006;16:1136–48. doi: 10.1101/gr.5402306. - DOI - PMC - PubMed
-
- Kermani BG. Artificial intelligence and global normalization methods for genotyping. 2008. http://www.patentstorm.us/patents/7467117/fulltext.html
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
