

Angiogenic functions of voltage-gated Na+ Channels in human endothelial cells: modulation of vascular endothelial growth factor (VEGF) signaling
- PMID: 21385874
- PMCID: PMC3089528
- DOI: 10.1074/jbc.M110.187559
Angiogenic functions of voltage-gated Na+ Channels in human endothelial cells: modulation of vascular endothelial growth factor (VEGF) signaling
Abstract
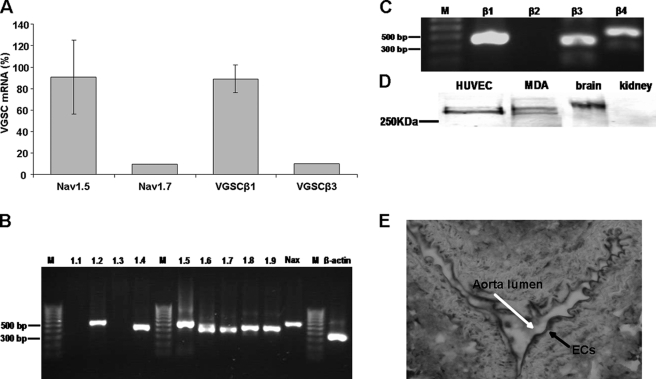
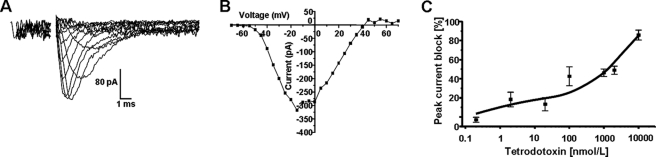
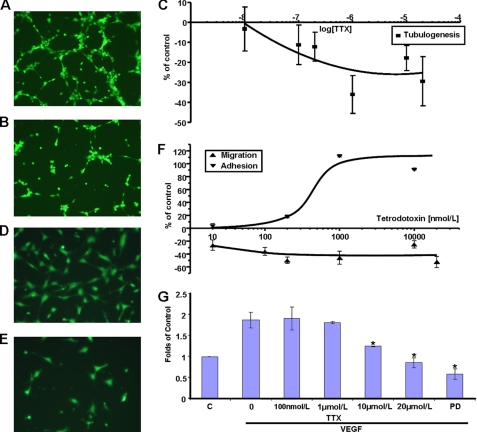
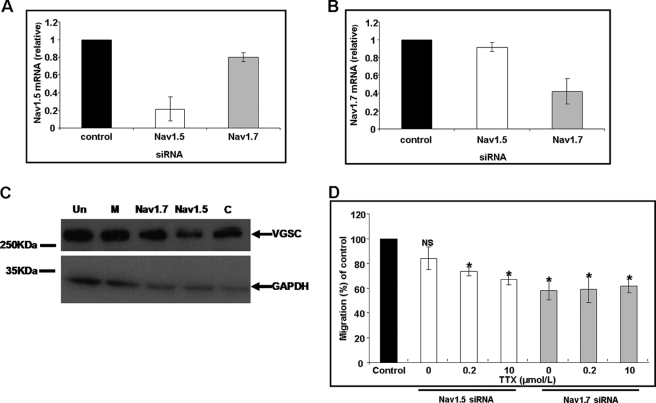
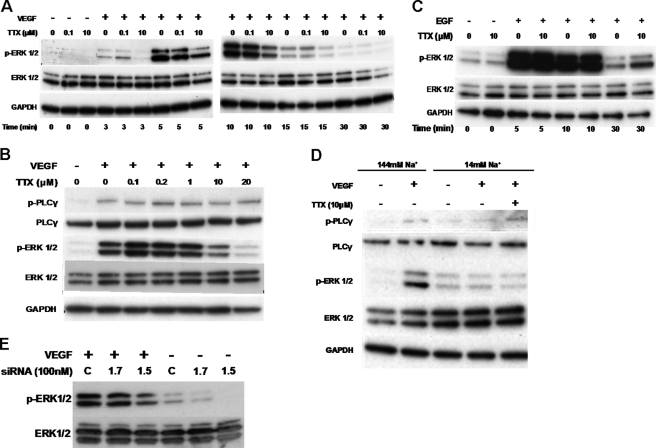
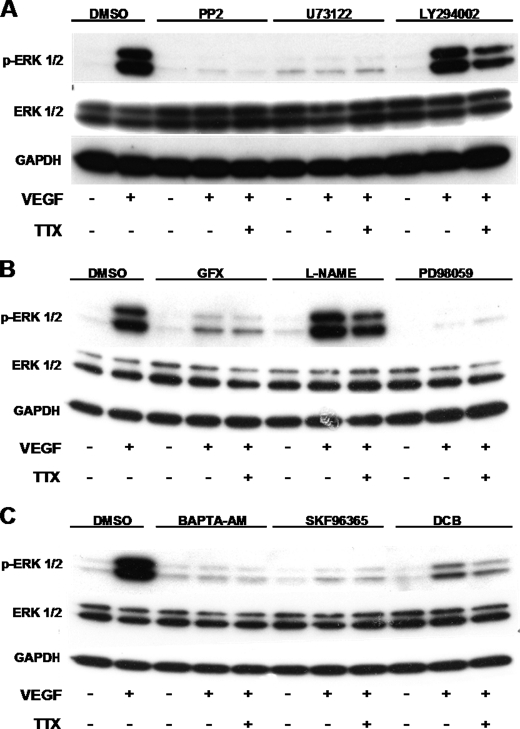
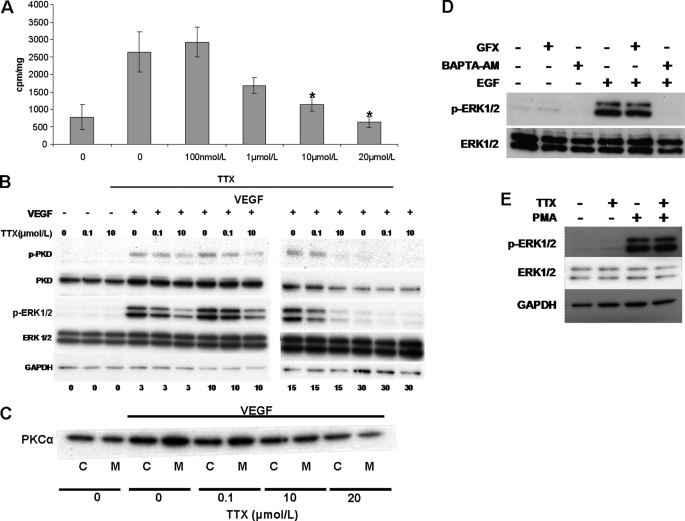
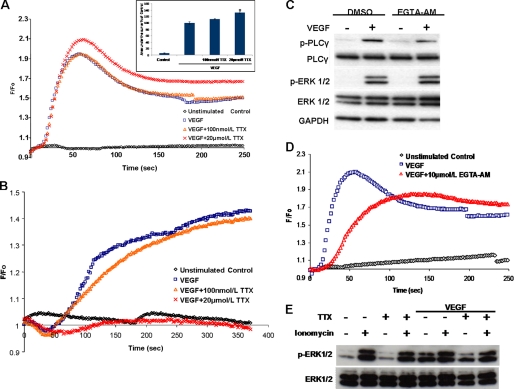
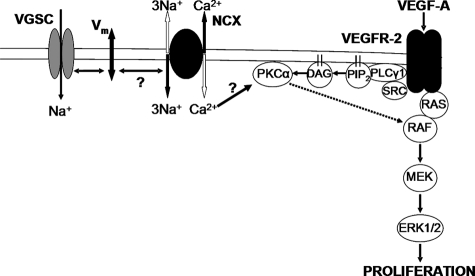
Voltage-gated sodium channel (VGSC) activity has previously been reported in endothelial cells (ECs). However, the exact isoforms of VGSCs present, their mode(s) of action, and potential role(s) in angiogenesis have not been investigated. The main aims of this study were to determine the role of VGSC activity in angiogenic functions and to elucidate the potentially associated signaling mechanisms using human umbilical vein endothelial cells (HUVECs) as a model system. Real-time PCR showed that the primary functional VGSC α- and β-subunit isoforms in HUVECs were Nav1.5, Nav1.7, VGSCβ1, and VGSCβ3. Western blots verified that VGSCα proteins were expressed in HUVECs, and immunohistochemistry revealed VGSCα expression in mouse aortic ECs in vivo. Electrophysiological recordings showed that the channels were functional and suppressed by tetrodotoxin (TTX). VGSC activity modulated the following angiogenic properties of HUVECs: VEGF-induced proliferation or chemotaxis, tubular differentiation, and substrate adhesion. Interestingly, different aspects of angiogenesis were controlled by the different VGSC isoforms based on TTX sensitivity and effects of siRNA-mediated gene silencing. Additionally, we show for the first time that TTX-resistant (TTX-R) VGSCs (Nav1.5) potentiate VEGF-induced ERK1/2 activation through the PKCα-B-RAF signaling axis. We postulate that this potentiation occurs through modulation of VEGF-induced HUVEC depolarization and [Ca(2+)](i). We conclude that VGSCs regulate multiple angiogenic functions and VEGF signaling in HUVECs. Our results imply that targeting VGSC expression/activity could be a novel strategy for controlling angiogenesis.
Figures
References
-
- Carmeliet P. (2005) Nature 438, 932–936 - PubMed
-
- Chen C. H., Walterscheid J. P. (2006) Circ. Res. 99, 787–789 - PubMed
-
- Isner J. M. (2002) Nature 415, 234–239 - PubMed
-
- Semenza G. L. (2006) Circ. Res. 98, 1115–1116 - PubMed
-
- Olsson A. K., Dimberg A., Kreuger J., Claesson-Welsh L. (2006) Nat. Rev. Mol. Cell Biol. 7, 359–371 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
