

Removing batch effects in analysis of expression microarray data: an evaluation of six batch adjustment methods
- PMID: 21386892
- PMCID: PMC3046121
- DOI: 10.1371/journal.pone.0017238
Removing batch effects in analysis of expression microarray data: an evaluation of six batch adjustment methods
Abstract
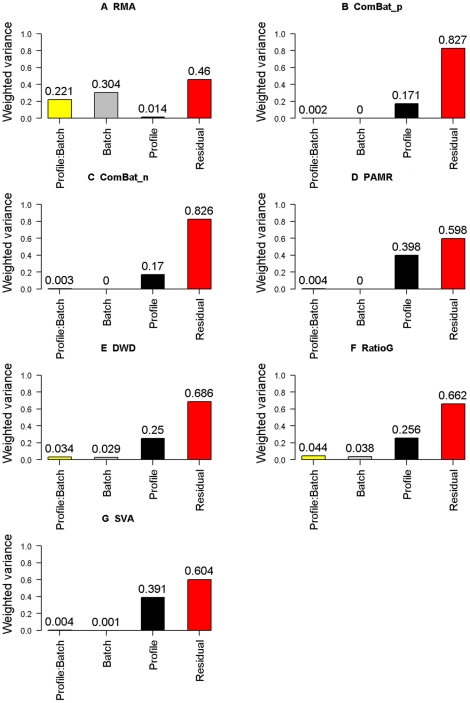
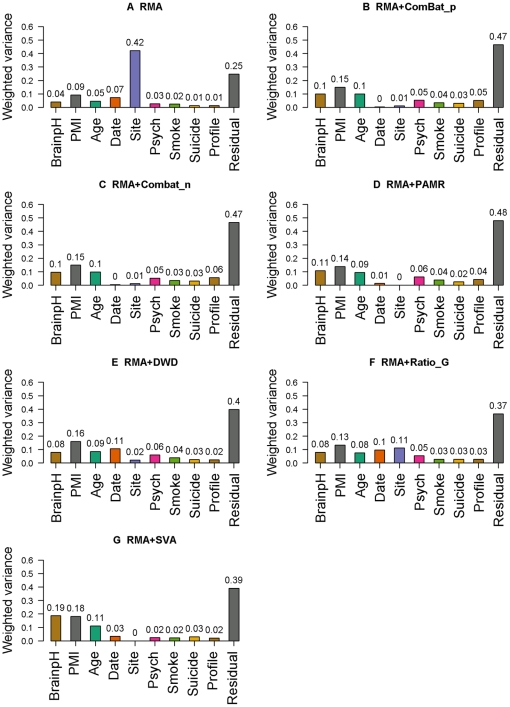
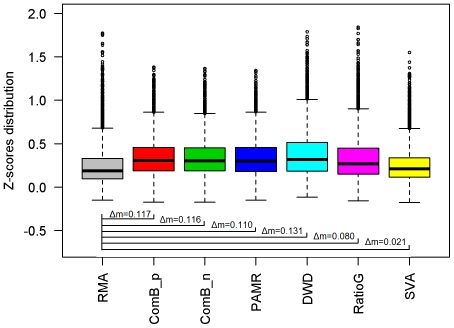
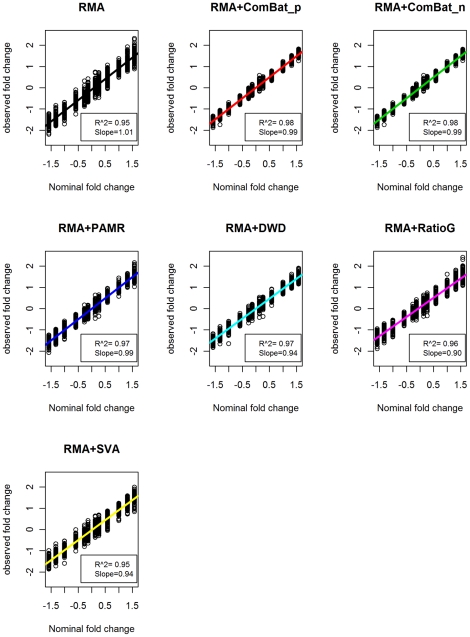
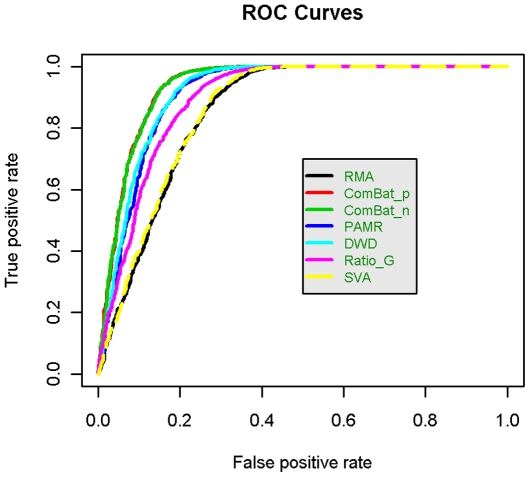
The expression microarray is a frequently used approach to study gene expression on a genome-wide scale. However, the data produced by the thousands of microarray studies published annually are confounded by "batch effects," the systematic error introduced when samples are processed in multiple batches. Although batch effects can be reduced by careful experimental design, they cannot be eliminated unless the whole study is done in a single batch. A number of programs are now available to adjust microarray data for batch effects prior to analysis. We systematically evaluated six of these programs using multiple measures of precision, accuracy and overall performance. ComBat, an Empirical Bayes method, outperformed the other five programs by most metrics. We also showed that it is essential to standardize expression data at the probe level when testing for correlation of expression profiles, due to a sizeable probe effect in microarray data that can inflate the correlation among replicates and unrelated samples.
Conflict of interest statement
Figures
References
-
- Brown PO, Botstein D. Exploring the new world of the genome with DNA microarrays. Nature Genetics. 1999;21:33–37. - PubMed
-
- Lockhart DJ, Dong HL, Byrne MC, Follettie MT, Gallo MV, et al. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nature Biotechnology. 1996;14:1675–1680. - PubMed
-
- Schena M, Shalon D, Davis RW, Brown PO. Quantitative Monitoring of Gene-Expression Patterns with a Complementary-DNA Microarray. Science. 1995;270:467–470. - PubMed
-
- Sims AH. Bioinformatics and breast cancer: what can high-throughput genomic approaches actually tell us? Journal of Clinical Pathology. 2009;62:879–885. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
