

ADAM12 induces estrogen-independence in breast cancer cells
- PMID: 21387162
- PMCID: PMC3381741
- DOI: 10.1007/s10549-011-1431-4
ADAM12 induces estrogen-independence in breast cancer cells
Abstract
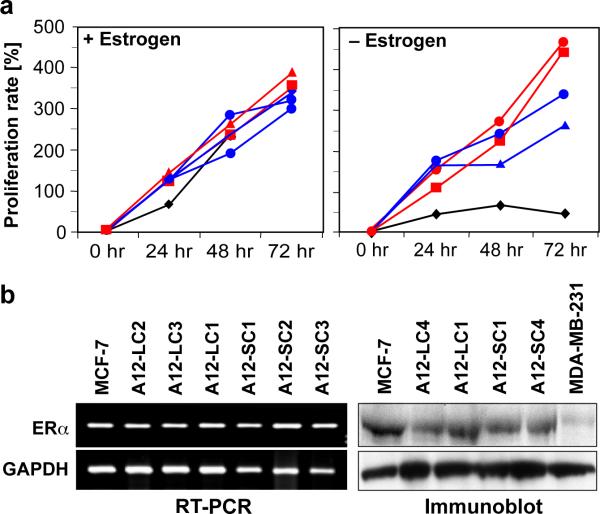
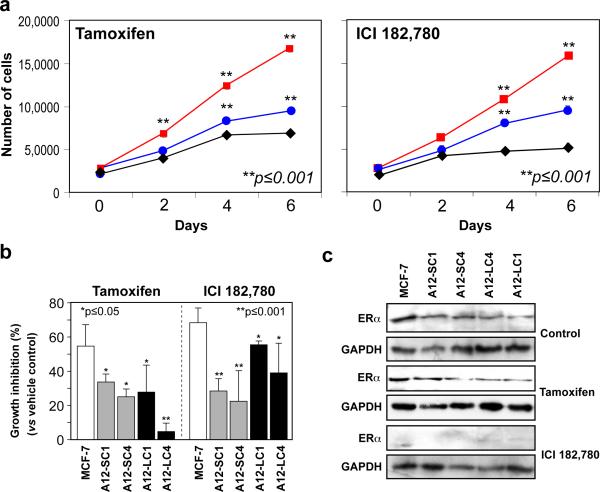
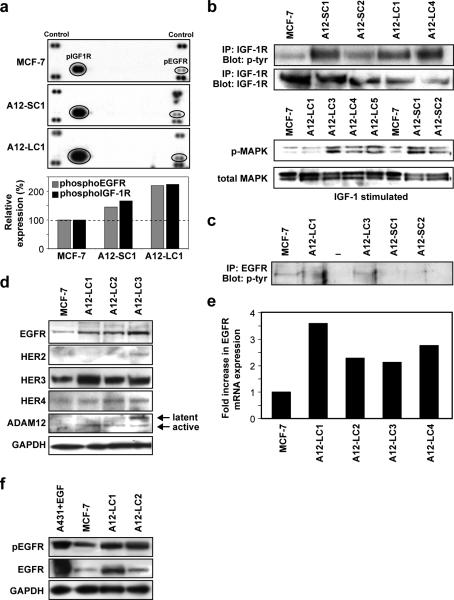
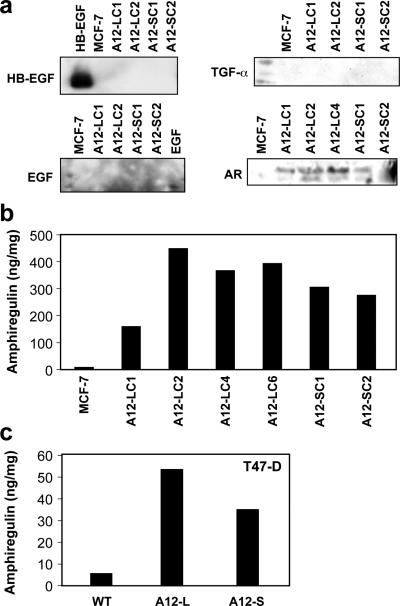
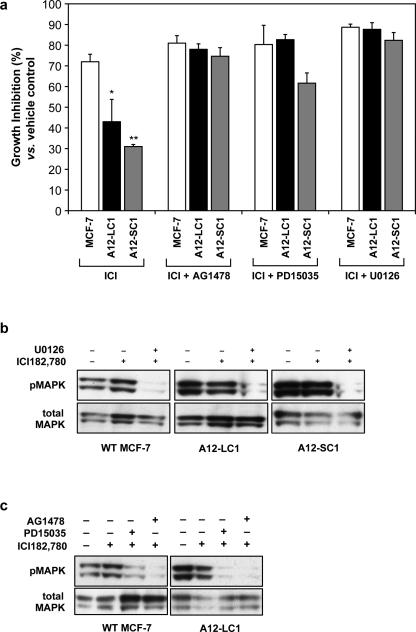
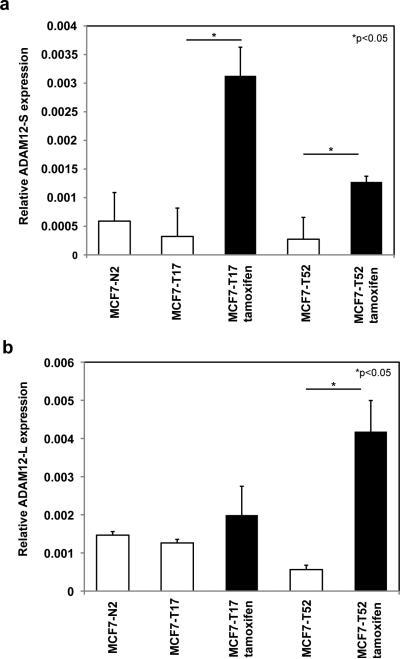
Antiestrogen therapy has been used successfully to prolong disease-free and overall survival of ER positive breast cancer patients. However, 50% of patients with ER+ tumors fail to respond to such therapy or eventually acquire resistance to endocrine therapy, resulting in tumor progression and mortality. It is imperative, therefore, to understand the mechanisms that lead to hormone refractory breast cancer in order to develop therapeutics that can modulate the resistance to antiestrogen therapy. The protease, ADAM12, can be detected in the urine of breast cancer patients and its levels correlate with disease status, stage, and cancer risk. Within the context of this study, the authors have investigated the role of the two distinct isoforms of ADAM12 in breast tumor cell proliferation and as potential mediators of endocrine resistance. Using stable clones of ADAM12-overexpressing MCF-7 cells, the authors analyzed proliferation rates of these ER+ breast tumor cells both in estrogen-depleted medium and in the presence of the antiestrogens, tamoxifen, and ICI 182,780. Acquired estrogen resistance in these cells was analyzed using phospho-RTK analysis. Upregulation and phosphorylation of proteins were detected via immunoprecipitation and immunoblotting. EGFR and MAPK inhibitors were used to explore the mechanism of acquired estrogen resistance in breast tumor cells. It was observed that overexpression of the two isoforms, transmembrane ADAM12-L, and secreted ADAM12-S, in breast tumor cells promoted estrogen-independent proliferation. In ADAM12-L-expressing cells, estrogen-independence was a direct result of increased EGFR expression and MAPK activation, whereas, the mechanism in ADAM12-S-expressing cells may be enhanced IGF-1R signaling. The importance of the EGFR signaling pathway in the estrogen-independent growth of ADAM12-L expressing cells was highlighted by the effect of EGFR inhibitors AG1478 and PD15035 or MAPK inhibitor U0126, each of which abolished the antiestrogen resistance in these cells. Taken together, these results demonstrate that ADAM12 isoforms confer a proliferative advantage to MCF-7 cells in the absence of estrogen stimulation, and suggest that downregulation of ADAM12 in combination with endocrine therapy may represent a useful pharmacological approach to breast cancer therapy.
Figures
Similar articles
-
The role of amphiregulin in exemestane-resistant breast cancer cells: evidence of an autocrine loop.Cancer Res. 2008 Apr 1;68(7):2259-65. doi: 10.1158/0008-5472.CAN-07-5544. Cancer Res. 2008. PMID: 18381432 Free PMC article.
-
Fulvestrant regulates epidermal growth factor (EGF) family ligands to activate EGF receptor (EGFR) signaling in breast cancer cells.Breast Cancer Res Treat. 2013 Jun;139(2):351-60. doi: 10.1007/s10549-013-2541-y. Epub 2013 May 18. Breast Cancer Res Treat. 2013. PMID: 23686416 Free PMC article.
-
Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer.J Natl Cancer Inst. 2004 Jun 16;96(12):926-35. doi: 10.1093/jnci/djh166. J Natl Cancer Inst. 2004. PMID: 15199112
-
Endocrine resistance: what do we know?Am Soc Clin Oncol Educ Book. 2013. doi: 10.14694/EdBook_AM.2013.33.e37. Am Soc Clin Oncol Educ Book. 2013. PMID: 23714450 Review.
-
Inhibition of erbB receptor (HER) tyrosine kinases as a strategy to abrogate antiestrogen resistance in human breast cancer.Clin Cancer Res. 2001 Dec;7(12 Suppl):4436s-4442s; discussion 4411s-4412s. Clin Cancer Res. 2001. PMID: 11916237 Review.
Cited by
-
A disintegrin and metalloproteinase-12 (ADAM12): function, roles in disease progression, and clinical implications.Biochim Biophys Acta. 2013 Oct;1830(10):4445-55. doi: 10.1016/j.bbagen.2013.05.011. Epub 2013 May 13. Biochim Biophys Acta. 2013. PMID: 23680494 Free PMC article. Review.
-
ADAM12 produced by tumor cells rather than stromal cells accelerates breast tumor progression.Mol Cancer Res. 2011 Nov;9(11):1449-61. doi: 10.1158/1541-7786.MCR-11-0100. Epub 2011 Aug 29. Mol Cancer Res. 2011. PMID: 21875931 Free PMC article.
-
Upregulation of ADAM12 Is Associated With a Poor Survival and Immune Cell Infiltration in Colon Adenocarcinoma.Front Oncol. 2021 Sep 16;11:729230. doi: 10.3389/fonc.2021.729230. eCollection 2021. Front Oncol. 2021. PMID: 34604068 Free PMC article.
-
ADAM12 transmembrane and secreted isoforms promote breast tumor growth: a distinct role for ADAM12-S protein in tumor metastasis.J Biol Chem. 2011 Jun 10;286(23):20758-68. doi: 10.1074/jbc.M110.216036. Epub 2011 Apr 14. J Biol Chem. 2011. PMID: 21493715 Free PMC article.
-
ADAM12 Is a Novel Regulator of Tumor Angiogenesis via STAT3 Signaling.Mol Cancer Res. 2017 Nov;15(11):1608-1622. doi: 10.1158/1541-7786.MCR-17-0188. Epub 2017 Aug 1. Mol Cancer Res. 2017. PMID: 28765266 Free PMC article.
References
-
- Ravdin PM, Cronin KA, Howlader N, Berg CD, Chlebowski RT, Feuer EJ, Edwards BK, Berry DA. The decrease in breast-cancer incidence in 2003 in the united states. N Engl J Med. 2007;356(16):1670–1674. doi:356/16/1670 [pii]10.1056/NEJMsr070105. - PubMed
-
- Osborne CK, Fuqua SA. Mechanisms of tamoxifen resistance. Breast Cancer Res Treat. 1994;32(1):49–55. - PubMed
-
- McCune K, Bhat-Nakshatri P, Thorat MA, Nephew KP, Badve S, Nakshatri H. Prognosis of hormone-dependent breast cancers: Implications of the presence of dysfunctional transcriptional networks activated by insulin via the immune transcription factor t-bet. Cancer Res. 70(2):685–696. doi:0008-5472.CAN-09-1530 [pii]10.1158/0008-5472.CAN-09-1530. - PMC - PubMed
-
- Frogne T, Benjaminsen RV, Sonne-Hansen K, Sorensen BS, Nexo E, Laenkholm AV, Rasmussen LM, Riese DJ, 2nd, de Cremoux P, Stenvang J, Lykkesfeldt AE. Activation of erbb3, egfr and erk is essential for growth of human breast cancer cell lines with acquired resistance to fulvestrant. Breast Cancer Res Treat. 2009;114(2):263–275. doi:10.1007/s10549-008-0011-8. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
