

SOS1 mutations in Noonan syndrome: molecular spectrum, structural insights on pathogenic effects, and genotype-phenotype correlations
- PMID: 21387466
- PMCID: PMC3118925
- DOI: 10.1002/humu.21492
SOS1 mutations in Noonan syndrome: molecular spectrum, structural insights on pathogenic effects, and genotype-phenotype correlations
Abstract
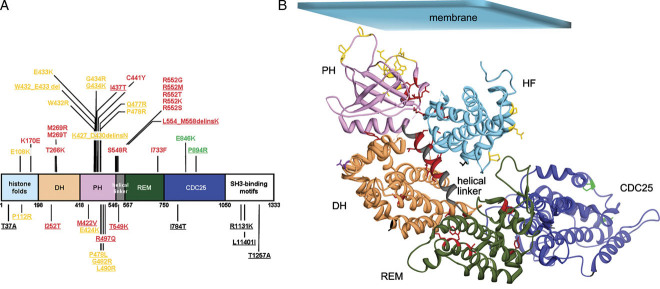
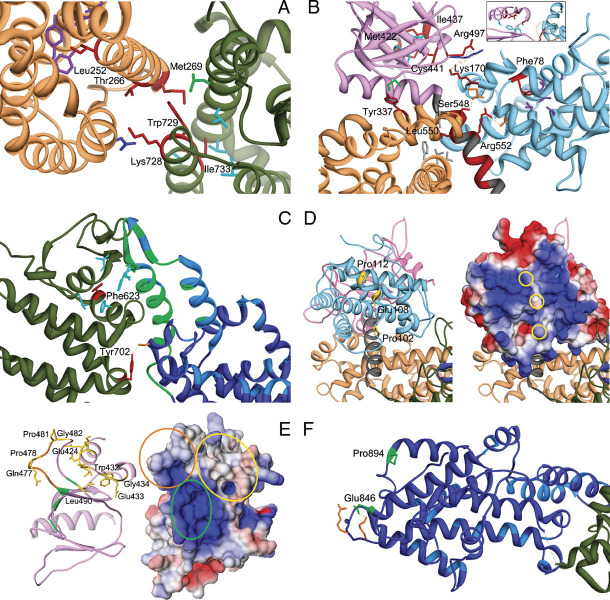
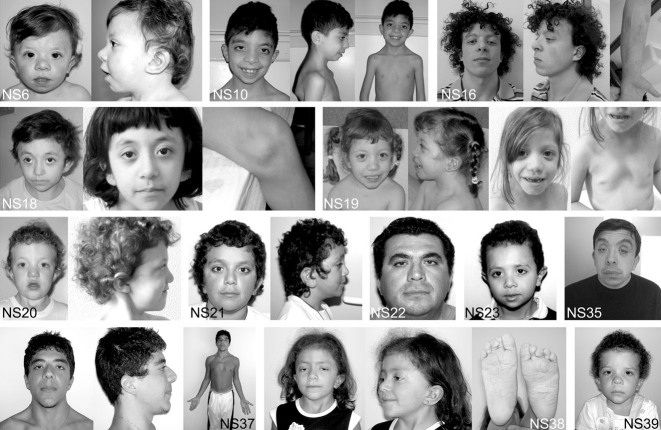
Noonan syndrome (NS) is among the most common nonchromosomal disorders affecting development and growth. NS is caused by aberrant RAS-MAPK signaling and is genetically heterogeneous, which explains, in part, the marked clinical variability documented for this Mendelian trait. Recently, we and others identified SOS1 as a major gene underlying NS. Here, we explored further the spectrum of SOS1 mutations and their associated phenotypic features. Mutation scanning of the entire SOS1 coding sequence allowed the identification of 33 different variants deemed to be of pathological significance, including 16 novel missense changes and in-frame indels. Various mutation clusters destabilizing or altering orientation of regions of the protein predicted to contribute structurally to the maintenance of autoinhibition were identified. Two previously unappreciated clusters predicted to enhance SOS1's recruitment to the plasma membrane, thus promoting a spatial reorientation of domains contributing to inhibition, were also recognized. Genotype-phenotype analysis confirmed our previous observations, establishing a high frequency of ectodermal anomalies and a low prevalence of cognitive impairment and reduced growth. Finally, mutation analysis performed on cohorts of individuals with nonsyndromic pulmonic stenosis, atrial septal defects, and ventricular septal defects excluded a major contribution of germline SOS1 lesions to the isolated occurrence of these cardiac anomalies.
© 2011 Wiley-Liss, Inc.
Figures
References
-
- Aoki Y, Niihori T, Narumi Y, Kure S, Matsubara Y. The RAS/MAPK syndromes: novel roles of the RAS pathway in human genetic disorders. Hum Mutat. 2008;29:992–1006. - PubMed
-
- Aronheim A, Engelberg D, Li N, al-Alawi N, Schlessinger J, Karin M. Membrane targeting of the nucleotide exchange factor Sos is sufficient for activating the Ras signaling pathway. Cell. 1994;78:949–961. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
