

A dystroglycan mutation associated with limb-girdle muscular dystrophy
- PMID: 21388311
- PMCID: PMC3071687
- DOI: 10.1056/NEJMoa1006939
A dystroglycan mutation associated with limb-girdle muscular dystrophy
Abstract
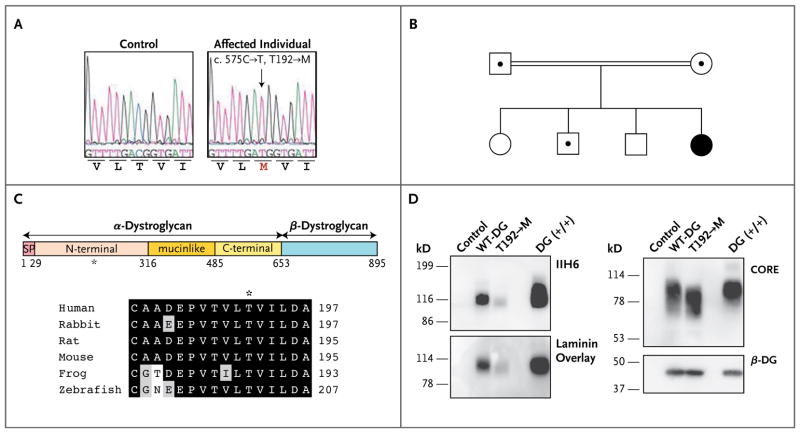
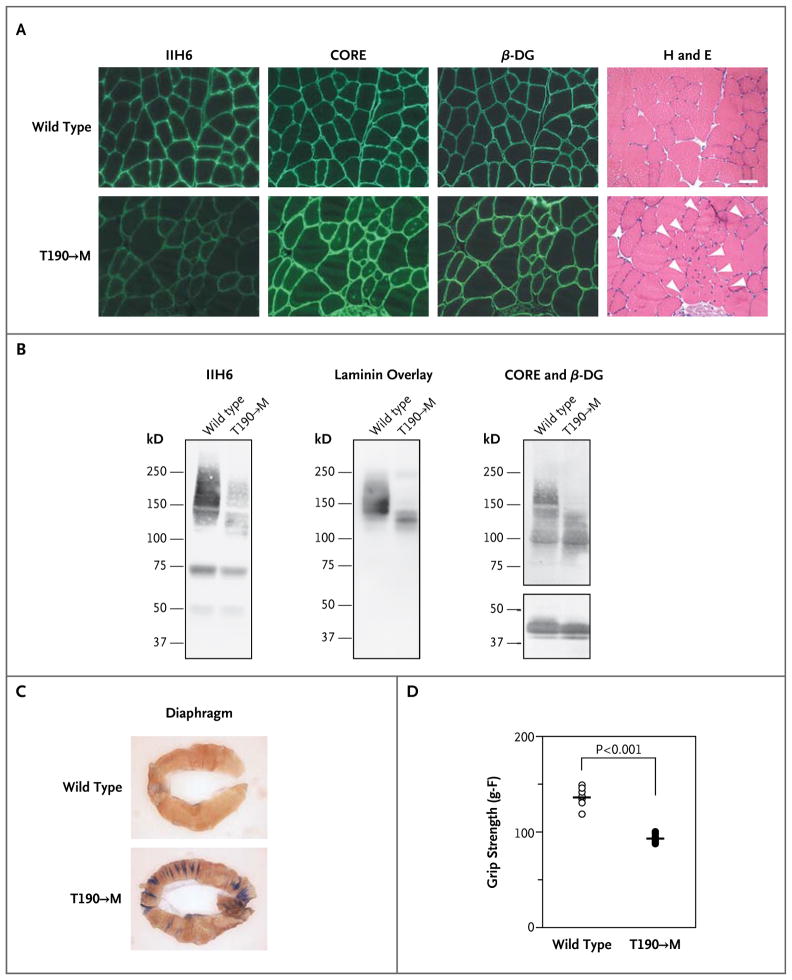
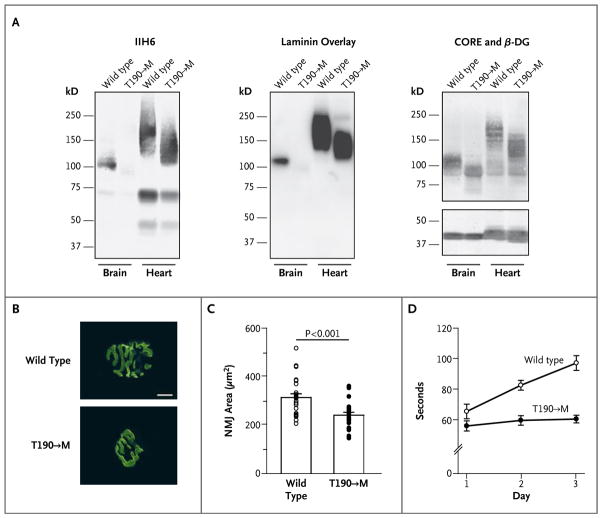
Dystroglycan, which serves as a major extracellular matrix receptor in muscle and the central nervous system, requires extensive O-glycosylation to function. We identified a dystroglycan missense mutation (Thr192→Met) in a woman with limb-girdle muscular dystrophy and cognitive impairment. A mouse model harboring this mutation recapitulates the immunohistochemical and neuromuscular abnormalities observed in the patient. In vitro and in vivo studies showed that the mutation impairs the receptor function of dystroglycan in skeletal muscle and brain by inhibiting the post-translational modification, mediated by the glycosyltransferase LARGE, of the phosphorylated O-mannosyl glycans on α-dystroglycan that is required for high-affinity binding to laminin.
Figures
References
-
- Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355:696–702. - PubMed
-
- Barresi R, Campbell KP. Dystroglycan: from biosynthesis to pathogenesis of human disease. J Cell Sci. 2006;119:199–207. - PubMed
-
- Michele DE, Barresi R, Kanagawa M, et al. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature. 2002;418:417–22. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials