

Secreted bacterial effectors that inhibit host protein synthesis are critical for induction of the innate immune response to virulent Legionella pneumophila
- PMID: 21390206
- PMCID: PMC3040669
- DOI: 10.1371/journal.ppat.1001289
Secreted bacterial effectors that inhibit host protein synthesis are critical for induction of the innate immune response to virulent Legionella pneumophila
Abstract
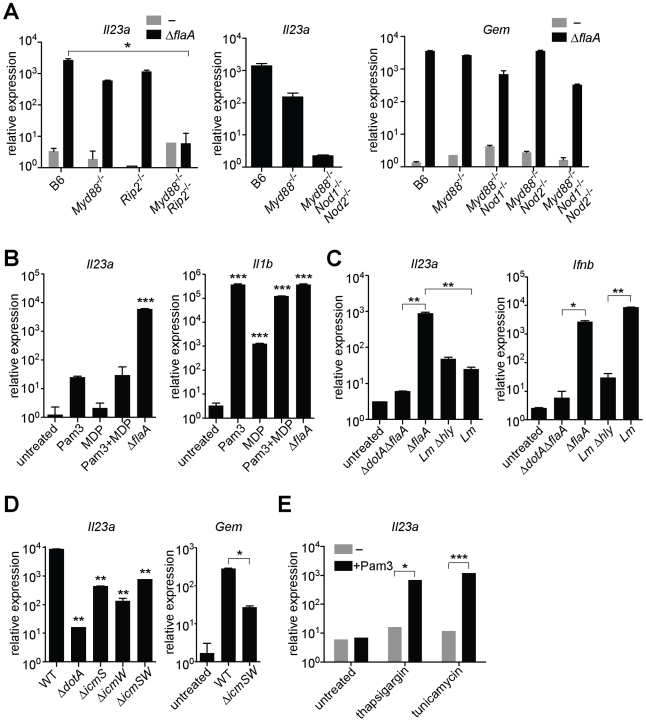
The intracellular bacterial pathogen Legionella pneumophila causes an inflammatory pneumonia called Legionnaires' Disease. For virulence, L. pneumophila requires a Dot/Icm type IV secretion system that translocates bacterial effectors to the host cytosol. L. pneumophila lacking the Dot/Icm system is recognized by Toll-like receptors (TLRs), leading to a canonical NF-κB-dependent transcriptional response. In addition, L. pneumophila expressing a functional Dot/Icm system potently induces unique transcriptional targets, including proinflammatory genes such as Il23a and Csf2. Here we demonstrate that this Dot/Icm-dependent response, which we term the effector-triggered response (ETR), requires five translocated bacterial effectors that inhibit host protein synthesis. Upon infection of macrophages with virulent L. pneumophila, these five effectors caused a global decrease in host translation, thereby preventing synthesis of IκB, an inhibitor of the NF-κB transcription factor. Thus, macrophages infected with wildtype L. pneumophila exhibited prolonged activation of NF-κB, which was associated with transcription of ETR target genes such as Il23a and Csf2. L. pneumophila mutants lacking the five effectors still activated TLRs and NF-κB, but because the mutants permitted normal IκB synthesis, NF-κB activation was more transient and was not sufficient to fully induce the ETR. L. pneumophila mutants expressing enzymatically inactive effectors were also unable to fully induce the ETR, whereas multiple compounds or bacterial toxins that inhibit host protein synthesis via distinct mechanisms recapitulated the ETR when administered with TLR ligands. Previous studies have demonstrated that the host response to bacterial infection is induced primarily by specific microbial molecules that activate TLRs or cytosolic pattern recognition receptors. Our results add to this model by providing a striking illustration of how the host immune response to a virulent pathogen can also be shaped by pathogen-encoded activities, such as inhibition of host protein synthesis.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures






References
-
- Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. - PubMed
-
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. - PubMed
-
- Medzhitov R. Innate immunity: quo vadis? Nat Immunol. 2010;11:551–553. - PubMed
-
- Chisholm ST, Coaker G, Day B, Staskawicz BJ. Host-microbe interactions: shaping the evolution of the plant immune response. Cell. 2006;124:803–814. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
