

Direct regulation of complex I by mitochondrial MEF2D is disrupted in a mouse model of Parkinson disease and in human patients
- PMID: 21393861
- PMCID: PMC3049386
- DOI: 10.1172/JCI43871
Direct regulation of complex I by mitochondrial MEF2D is disrupted in a mouse model of Parkinson disease and in human patients
Abstract
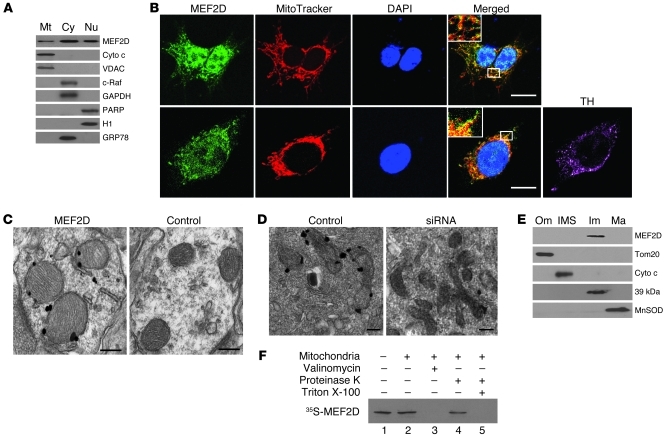
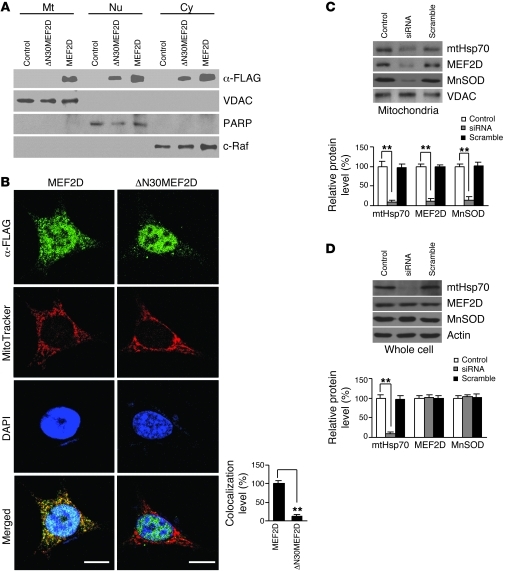
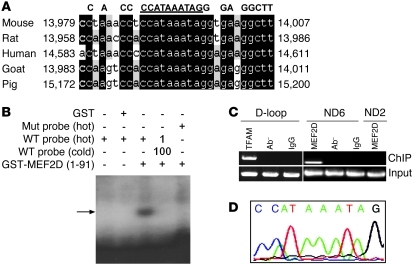
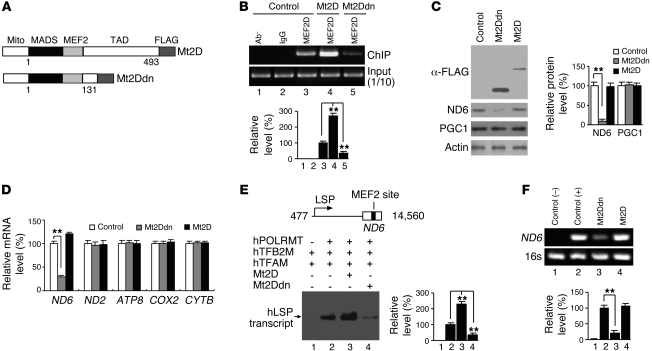
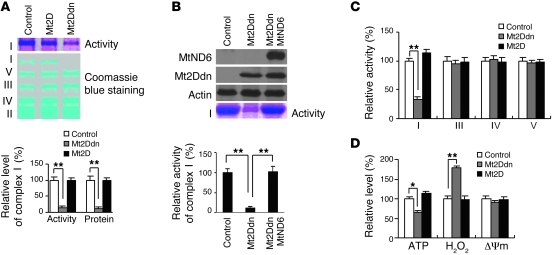
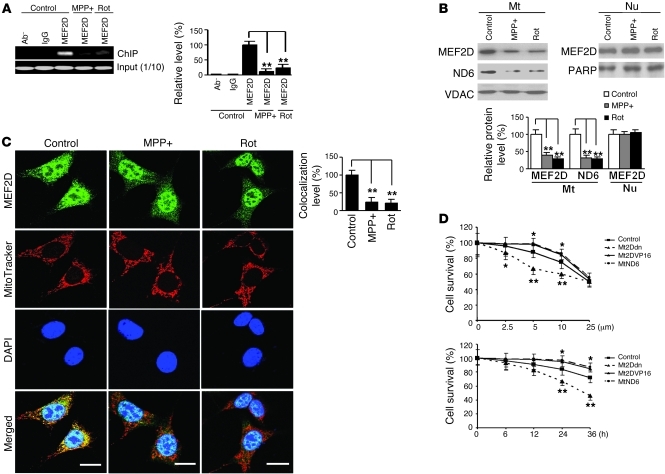
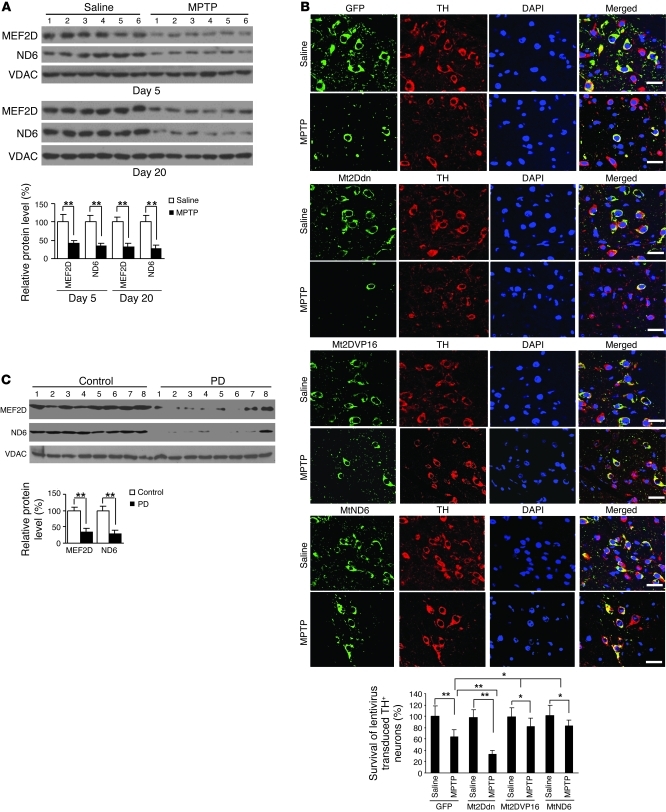
The transcription factors in the myocyte enhancer factor 2 (MEF2) family play important roles in cell survival by regulating nuclear gene expression. Here, we report that MEF2D is present in rodent neuronal mitochondria, where it can regulate the expression of a gene encoded within mitochondrial DNA (mtDNA). Immunocytochemical, immunoelectron microscopic, and biochemical analyses of rodent neuronal cells showed that a portion of MEF2D was targeted to mitochondria via an N-terminal motif and the chaperone protein mitochondrial heat shock protein 70 (mtHsp70). MEF2D bound to a MEF2 consensus site in the region of the mtDNA that contained the gene NADH dehydrogenase 6 (ND6), which encodes an essential component of the complex I enzyme of the oxidative phosphorylation system; MEF2D binding induced ND6 transcription. Blocking MEF2D function specifically in mitochondria decreased complex I activity, increased cellular H(2)O(2) level, reduced ATP production, and sensitized neurons to stress-induced death. Toxins known to affect complex I preferentially disrupted MEF2D function in a mouse model of Parkinson disease (PD). In addition, mitochondrial MEF2D and ND6 levels were decreased in postmortem brain samples of patients with PD compared with age-matched controls. Thus, direct regulation of complex I by mitochondrial MEF2D underlies its neuroprotective effects, and dysregulation of this pathway may contribute to PD.
Figures
References
-
- Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125(7):1241–1252. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
