

Enhanced efficacy of enzyme replacement therapy in Pompe disease through mannose-6-phosphate receptor expression in skeletal muscle
- PMID: 21397538
- PMCID: PMC3101281
- DOI: 10.1016/j.ymgme.2011.02.006
Enhanced efficacy of enzyme replacement therapy in Pompe disease through mannose-6-phosphate receptor expression in skeletal muscle
Abstract
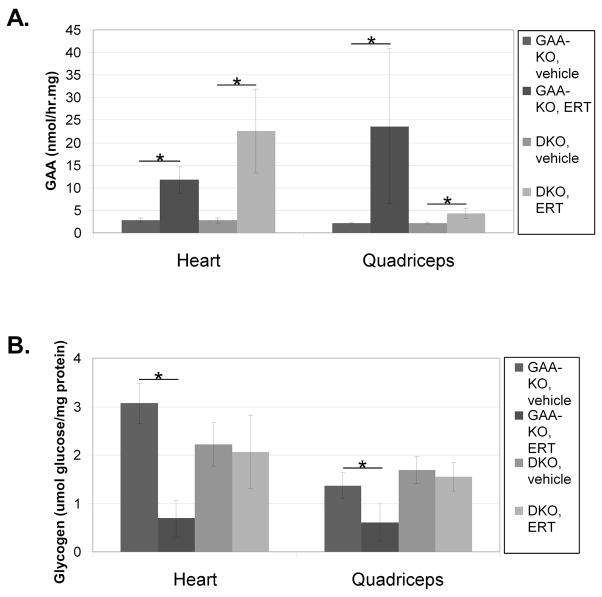
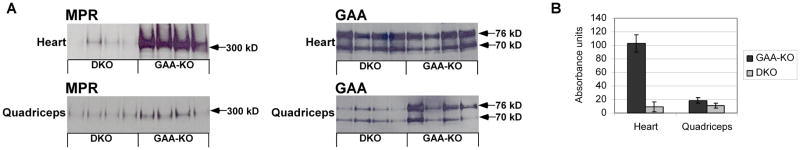
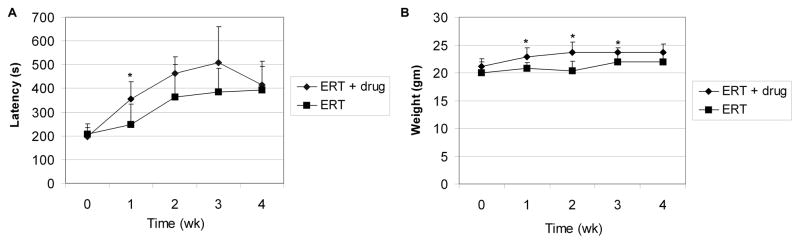
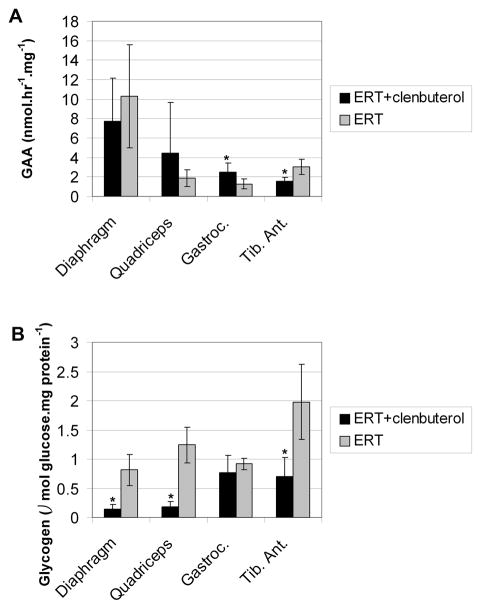
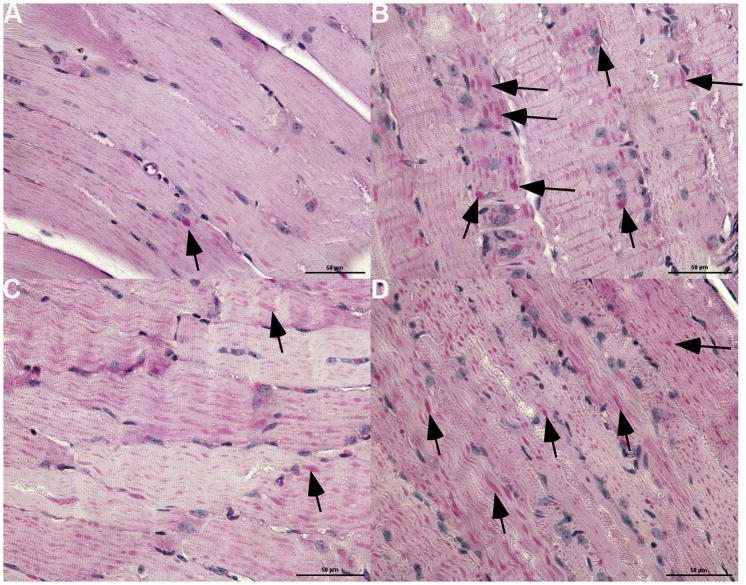
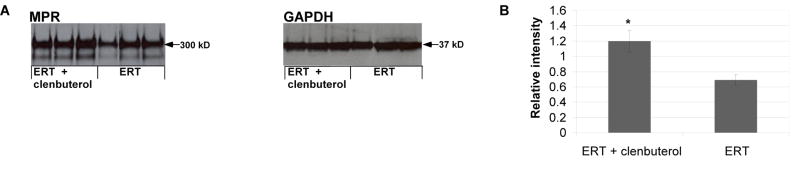
Enzyme replacement therapy (ERT) with acid α-glucosidase has become available for Pompe disease; however, the response of skeletal muscle, as opposed to the heart, has been attenuated. The poor response of skeletal muscle has been attributed to the low abundance of the cation-independent mannose-6-phosphate receptor (CI-MPR) in skeletal muscle compared to heart. To further understand the role of CI-MPR in Pompe disease, muscle-specific CI-MPR conditional knockout (KO) mice were crossed with GAA-KO (Pompe disease) mice. We evaluated the impact of CI-MPR-mediated uptake of GAA by evaluating ERT in CI-MPR-KO/GAA-KO (double KO) mice. The essential role of CI-MPR was emphasized by the lack of efficacy of ERT as demonstrated by markedly reduced biochemical correction of GAA deficiency and of glycogen accumulations in double KO mice, in comparison with the administration of the same therapeutic doses in GAA-KO mice. Clenbuterol, a selective β(2)-agonist, enhanced the CI-MPR expression in skeletal tissue and also increased efficacy from GAA therapy, thereby confirming the key role of CI-MPR with regard to enzyme replacement therapy in Pompe disease. Biochemical correction improved in both muscle and non-muscle tissues, indicating that therapy could be similarly enhanced in other lysosomal storage disorders. In summary, enhanced CI-MPR expression might improve the efficacy of enzyme replacement therapy in Pompe disease through enhancing receptor-mediated uptake of GAA.
Copyright © 2011 Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures
References
-
- Hirschhorn R, Reuser AJJ. Glycogen Storage Disease Type II: Acid α-Glucosidase (Acid Maltase) Deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Basis for Inherited Disease. McGraw-Hill; New York: 2001.
-
- Kishnani PS, Corzo D, Nicolino M, Byrne B, Mandel H, Hwu WL, Leslie N, Levine J, Spencer C, Mcdonald M, Li J, Dumontier J, Halberthal M, Chien YH, Hopkin R, Vijayaraghavan S, Gruskin D, Bartholomew D, Van Der PA, Clancy JP, Parini R, Morin G, Beck M, De La Gastine GS, Jokic M, Thurberg B, Richards S, Bali D, Davison M, Worden MA, Chen YT, Wraith JE. Recombinant human acid {alpha}-glucosidase: Major clinical benefits in infantile-onset Pompe disease. Neurology. 2007;68:99–109. - PubMed
-
- Strothotte S, Strigl-Pill N, Grunert B, Kornblum C, Eger K, Wessig C, Deschauer M, Breunig F, Glocker FX, Vielhaber S, Brejova A, Hilz M, Reiners K, Muller-Felber W, Mengel E, Spranger M, Schoser B. Enzyme replacement therapy with alglucosidase alfa in 44 patients with late-onset glycogen storage disease type 2: 12-month results of an observational clinical trial. J Neurol. 2010;257:91–97. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
