

Academic cross-fertilization by public screening yields a remarkable class of protein phosphatase methylesterase-1 inhibitors
- PMID: 21398589
- PMCID: PMC3084096
- DOI: 10.1073/pnas.1015248108
Academic cross-fertilization by public screening yields a remarkable class of protein phosphatase methylesterase-1 inhibitors
Abstract
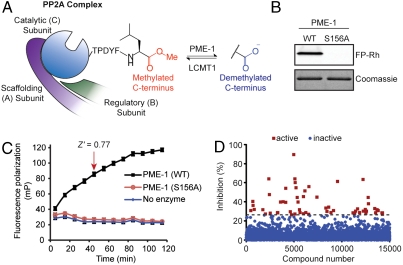
National Institutes of Health (NIH)-sponsored screening centers provide academic researchers with a special opportunity to pursue small-molecule probes for protein targets that are outside the current interest of, or beyond the standard technologies employed by, the pharmaceutical industry. Here, we describe the outcome of an inhibitor screen for one such target, the enzyme protein phosphatase methylesterase-1 (PME-1), which regulates the methylesterification state of protein phosphatase 2A (PP2A) and is implicated in cancer and neurodegeneration. Inhibitors of PME-1 have not yet been described, which we attribute, at least in part, to a dearth of substrate assays compatible with high-throughput screening. We show that PME-1 is assayable by fluorescence polarization-activity-based protein profiling (fluopol-ABPP) and use this platform to screen the 300,000+ member NIH small-molecule library. This screen identified an unusual class of compounds, the aza-β-lactams (ABLs), as potent (IC(50) values of approximately 10 nM), covalent PME-1 inhibitors. Interestingly, ABLs did not derive from a commercial vendor but rather an academic contribution to the public library. We show using competitive-ABPP that ABLs are exquisitely selective for PME-1 in living cells and mice, where enzyme inactivation leads to substantial reductions in demethylated PP2A. In summary, we have combined advanced synthetic and chemoproteomic methods to discover a class of ABL inhibitors that can be used to selectively perturb PME-1 activity in diverse biological systems. More generally, these results illustrate how public screening centers can serve as hubs to create spontaneous collaborative opportunities between synthetic chemistry and chemical biology labs interested in creating first-in-class pharmacological probes for challenging protein targets.
Conflict of interest statement
The authors declare no conflict of interest.
Figures





References
-
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. - PubMed
-
- Shi Y. Serine/threonine phosphatases: Mechanism through structure. Cell. 2009;139:468–484. - PubMed
-
- Oliver CJ, Shenolikar S. Physiologic importance of protein phosphatase inhibitors. Front Biosci. 1998;3:D961–972. - PubMed
-
- Kalhor HR, Luk K, Ramos A, Zobel-Thropp P, Clarke S. Protein phosphatase methyltransferase 1 (Ppm1p) is the sole activity responsible for modification of the major forms of protein phosphatase 2A in yeast. Arch Biochem Biophys. 2001;395:239–245. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
