

A novel method of characterizing genetic sequences: genome space with biological distance and applications
- PMID: 21399690
- PMCID: PMC3047556
- DOI: 10.1371/journal.pone.0017293
A novel method of characterizing genetic sequences: genome space with biological distance and applications
Erratum in
- PLoS One. 2011;6(3). doi: 10.1371/annotation/22351496-73dc-4205-9d9a-95a821ae74ca doi: 10.1371/annotation/22351496-73dc-4205-9d9a-95a821ae74ca
Abstract
Background: Most existing methods for phylogenetic analysis involve developing an evolutionary model and then using some type of computational algorithm to perform multiple sequence alignment. There are two problems with this approach: (1) different evolutionary models can lead to different results, and (2) the computation time required for multiple alignments makes it impossible to analyse the phylogeny of a whole genome. This motivates us to create a new approach to characterize genetic sequences.
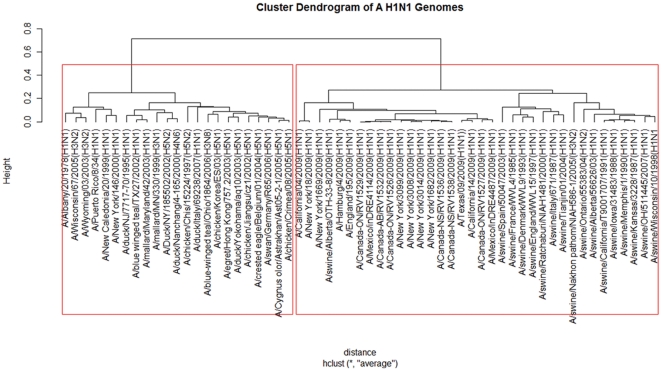
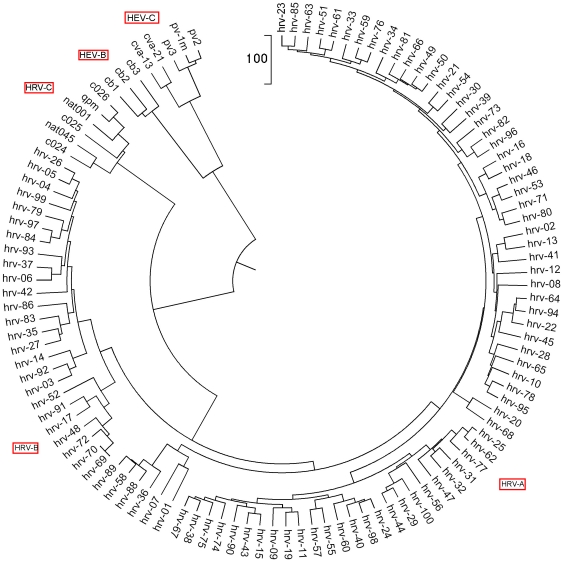
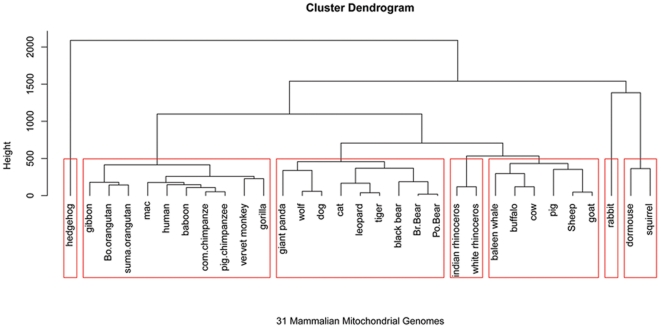
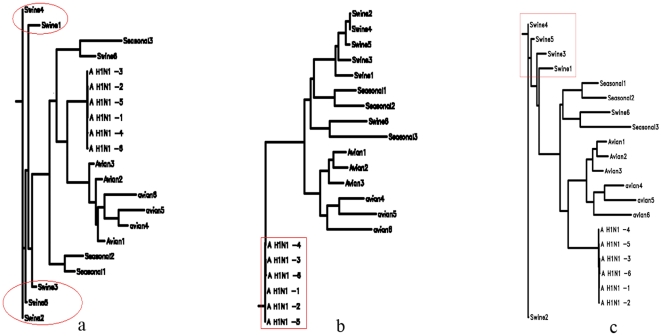
Methodology: To each DNA sequence, we associate a natural vector based on the distributions of nucleotides. This produces a one-to-one correspondence between the DNA sequence and its natural vector. We define the distance between two DNA sequences to be the distance between their associated natural vectors. This creates a genome space with a biological distance which makes global comparison of genomes with same topology possible. We use our proposed method to analyze the genomes of the new influenza A (H1N1) virus, human rhinoviruses (HRV) and mammalian mitochondrial. The result shows that a triple-reassortant swine virus circulating in North America and the Eurasian swine virus belong to the lineage of the influenza A (H1N1) virus. For the HRV and mammalian mitochondrial genomes, the results coincide with biologists' analyses.
Conclusions: Our approach provides a powerful new tool for analyzing and annotating genomes and their phylogenetic relationships. Whole or partial genomes can be handled more easily and more quickly than using multiple alignment methods. Once a genome space has been constructed, it can be stored in a database. There is no need to reconstruct the genome space for subsequent applications, whereas in multiple alignment methods, realignment is needed to add new sequences. Furthermore, one can make a global comparison of all genomes simultaneously, which no other existing method can achieve.
Conflict of interest statement
Figures
References
-
- Amano K, Nakamura H. Self-organizing clustering: a novel non-hierarchical method for clustering large amount of DNA sequences. Genome Inform. 2003;14:575–576.
-
- Emrich SJ, Kalyanaraman A, Aluru S. Aluru S, editor. Algorithms for large-scale clustering and assembly of biological sequence data. Handbook of Computational Molecular Biology. 2006. pp. 13.1–13.30.
-
- Waterman SM. Introduction to computational biology: maps, sequences and genomes. Boca Raton: Chapman & Hall/CRC Press; 1995. 431
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
