

Voltage-gated potassium channel KCNV2 (Kv8.2) contributes to epilepsy susceptibility
- PMID: 21402906
- PMCID: PMC3069171
- DOI: 10.1073/pnas.1017539108
Voltage-gated potassium channel KCNV2 (Kv8.2) contributes to epilepsy susceptibility
Abstract
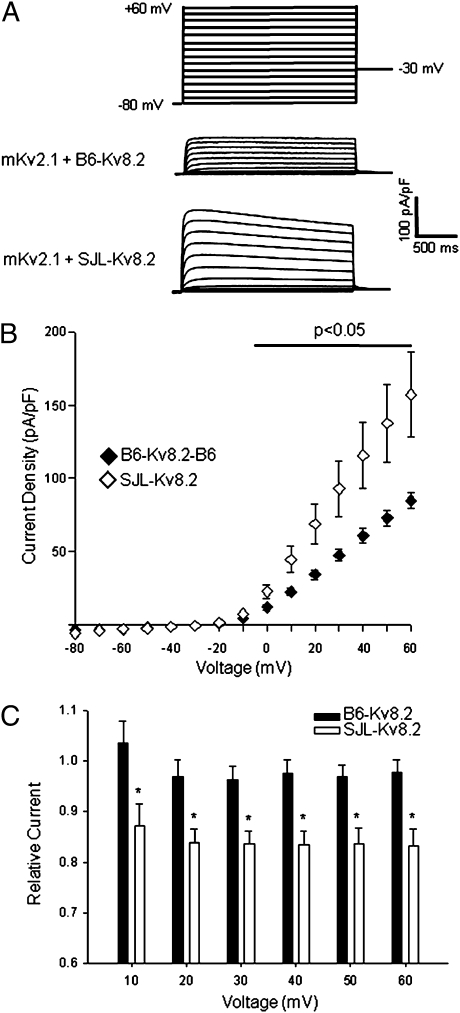
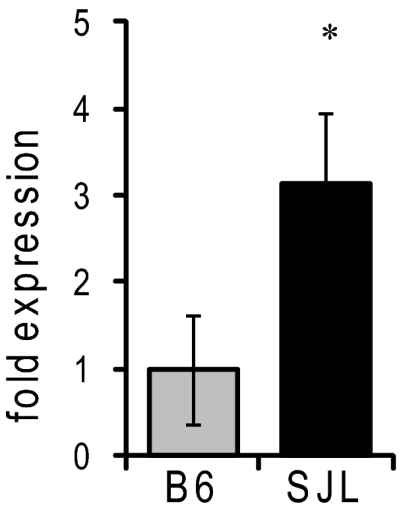
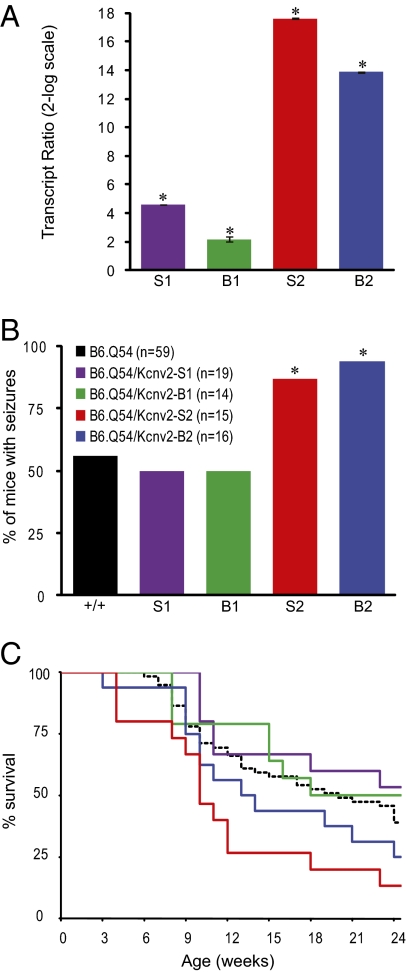
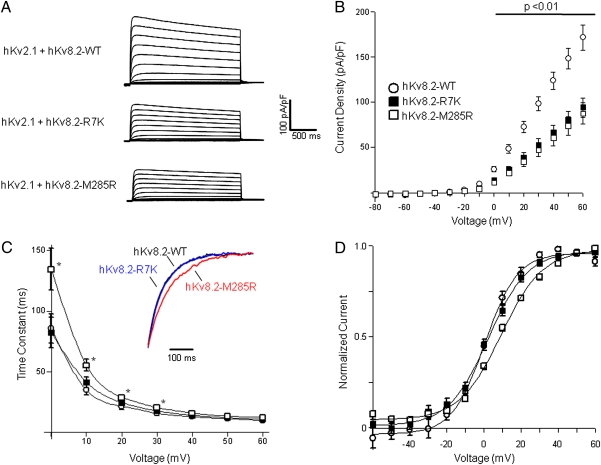
Mutations in voltage-gated ion channels are responsible for several types of epilepsy. Genetic epilepsies often exhibit variable severity in individuals with the same mutation, which may be due to variation in genetic modifiers. The Scn2a(Q54) transgenic mouse model has a sodium channel mutation and exhibits epilepsy with strain-dependent severity. We previously mapped modifier loci that influence Scn2a(Q54) phenotype severity and identified Kcnv2, encoding the voltage-gated potassium channel subunit Kv8.2, as a candidate modifier. In this study, we demonstrate a threefold increase in hippocampal Kcnv2 expression associated with more severe epilepsy. In vivo exacerbation of the phenotype by Kcnv2 transgenes supports its identification as an epilepsy modifier. The contribution of KCNV2 to human epilepsy susceptibility is supported by identification of two nonsynonymous variants in epilepsy patients that alter function of Kv2.1/Kv8.2 heterotetrameric potassium channels. Our results demonstrate that altered potassium subunit function influences epilepsy susceptibility and implicate Kcnv2 as an epilepsy gene.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Hauser WA, Annegers JF, Kurland LT. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935-1984. Epilepsia. 1993;34:453–468. - PubMed
-
- Leonardi M, Ustun TB. The global burden of epilepsy. Epilepsia. 2002;43(Suppl 6):21–25. - PubMed
-
- World Health Organization. Atlas: Epilepsy Care in the World. Geneva: WHO Press; 2005.
-
- Turnbull J, et al. Sacred disease secrets revealed: the genetics of human epilepsy. Hum Mol Genet. 2005;14(Spec No 2):2495–2500. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
