

Mitochondrial fusion/fission, transport and autophagy in Parkinson's disease: when mitochondria get nasty
- PMID: 21403911
- PMCID: PMC3043324
- DOI: 10.4061/2011/767230
Mitochondrial fusion/fission, transport and autophagy in Parkinson's disease: when mitochondria get nasty
Abstract
Understanding the molecular basis of Parkinson's disease (PD) has proven to be a major challenge in the field of neurodegenerative diseases. Although several hypotheses have been proposed to explain the molecular mechanisms underlying the pathogenesis of PD, a growing body of evidence has highlighted the role of mitochondrial dysfunction and the disruption of the mechanisms of mitochondrial dynamics in PD and other parkinsonian disorders. In this paper, we comment on the recent advances in how changes in the mitochondrial function and mitochondrial dynamics (fusion/fission, transport, and clearance) contribute to neurodegeneration, specifically focusing on PD. We also evaluate the current controversies in those issues and discuss the role of fusion/fission dynamics in the mitochondrial lifecycle and maintenance. We propose that cellular demise and neurodegeneration in PD are due to the interplay between mitochondrial dysfunction, mitochondrial trafficking disruption, and impaired autophagic clearance.
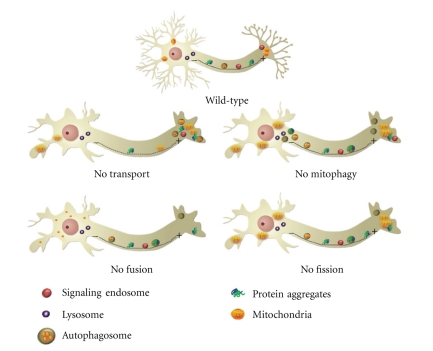
Figures

References
-
- Forno LS. Neuropathology of Parkinson’s disease. Journal of Neuropathology and Experimental Neurology. 1996;55(3):259–272. - PubMed
-
- Beal MF. Mitochondria, oxidative damage, and inflammation in Parkinson’s disease. Annals of the New York Academy of Sciences. 2003;991:120–131. - PubMed
-
- Schapira AH. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. The Lancet Neurology. 2008;7(1):97–109. - PubMed
-
- Cardoso SM, Esteves AR, Arduíno DM, Domingues AF, Oliveira CR. The crucial role of mitochondria in Parkinson’s disease. Recent Research Developments in Neuroscience. 2009;3:43–84.
LinkOut - more resources
Full Text Sources
Other Literature Sources
