

Novel mutations affecting LRP5 splicing in patients with osteoporosis-pseudoglioma syndrome (OPPG)
- PMID: 21407258
- PMCID: PMC3172925
- DOI: 10.1038/ejhg.2011.42
Novel mutations affecting LRP5 splicing in patients with osteoporosis-pseudoglioma syndrome (OPPG)
Abstract
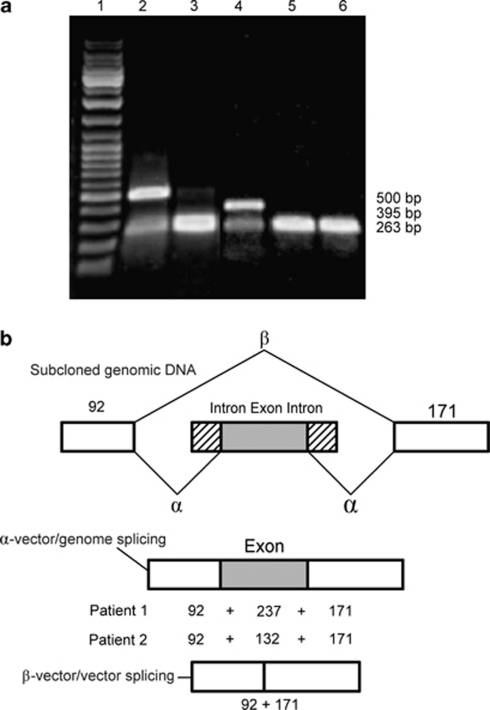
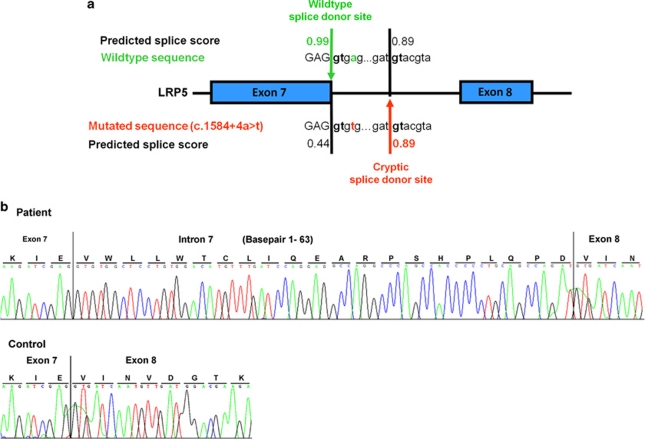
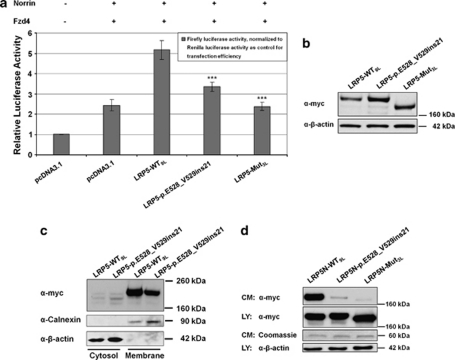
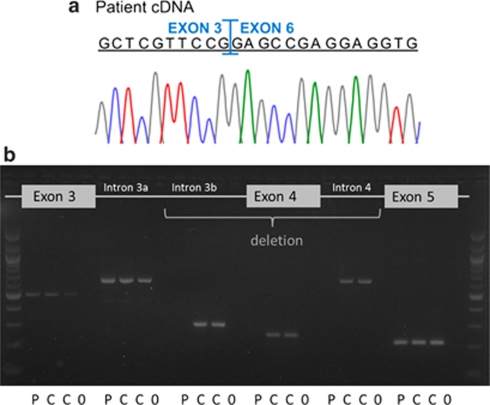
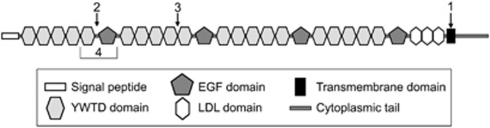
Osteoporosis-pseudoglioma sydrome (OPPG) is an autosomal recessive disorder with early-onset severe osteoporosis and blindness, caused by biallelic loss-of-function mutations in the low-density lipoprotein receptor-related protein 5 (LRP5) gene. Heterozygous carriers exhibit a milder bone phenotype. Only a few splice mutations in LRP5 have been published. We present clinical and genetic data for four patients with novel LRP5 mutations, three of which affect splicing. Patients were evaluated clinically and by radiography and bone densitometry. Genetic screening of LRP5 was performed on the basis of the clinical diagnosis of OPPG. Splice aberrances were confirmed by cDNA sequencing or exon trapping. The effect of one splice mutation on LRP5 protein function was studied. A novel splice-site mutation c.1584+4A>T abolished the donor splice site of exon 7 and activated a cryptic splice site, which led to an in-frame insertion of 21 amino acids (p.E528_V529ins21). Functional studies revealed severely impaired signal transduction presumably caused by defective intracellular transport of the mutated receptor. Exon trapping was used on two samples to confirm that splice-site mutations c.4112-2A>G and c.1015+1G>T caused splicing-out of exons 20 and 5, respectively. One patient carried a homozygous deletion of exon 4 causing the loss of exons 4 and 5, as demonstrated by cDNA analysis. Our results broaden the spectrum of mutations in LRP5 and provide the first functional data on splice aberrations.
Figures
References
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
