

Genomic inflation factors under polygenic inheritance
- PMID: 21407268
- PMCID: PMC3137506
- DOI: 10.1038/ejhg.2011.39
Genomic inflation factors under polygenic inheritance
Abstract
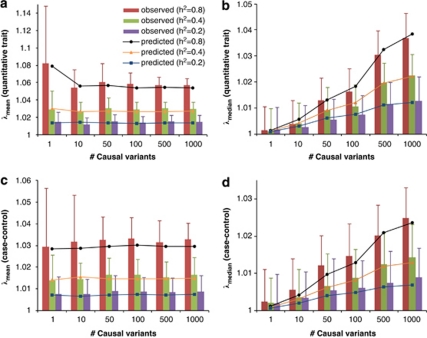
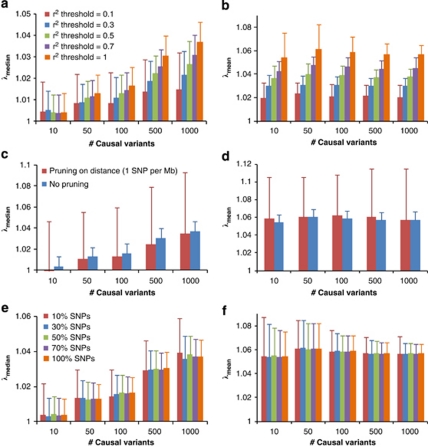
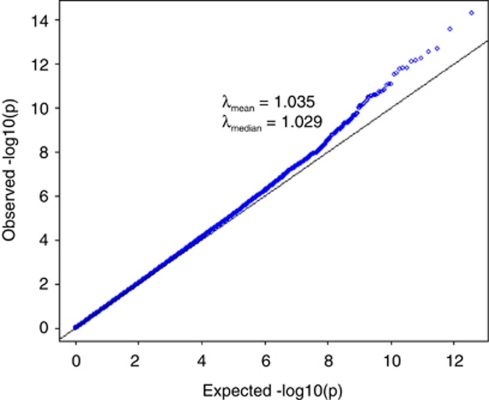
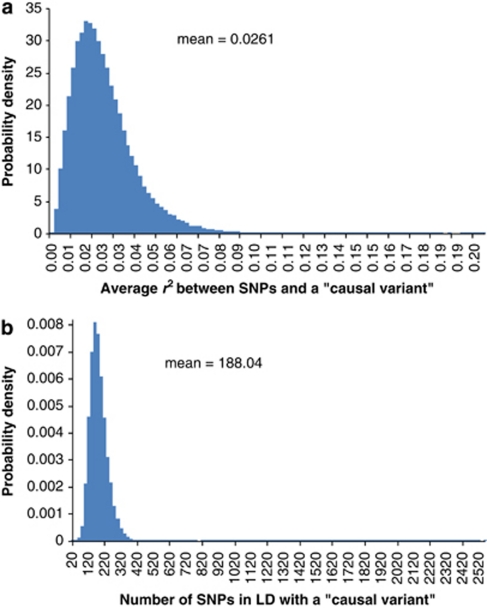
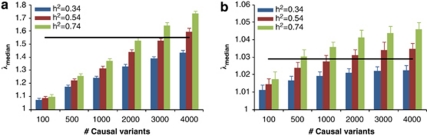
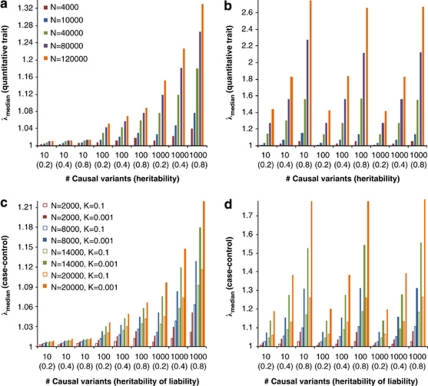
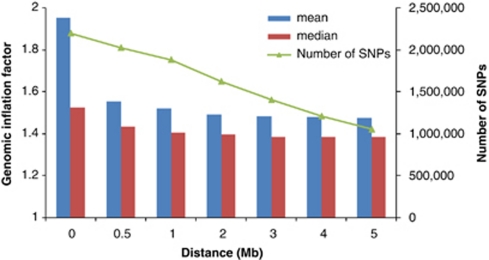
Population structure, including population stratification and cryptic relatedness, can cause spurious associations in genome-wide association studies (GWAS). Usually, the scaled median or mean test statistic for association calculated from multiple single-nucleotide-polymorphisms across the genome is used to assess such effects, and 'genomic control' can be applied subsequently to adjust test statistics at individual loci by a genomic inflation factor. Published GWAS have clearly shown that there are many loci underlying genetic variation for a wide range of complex diseases and traits, implying that a substantial proportion of the genome should show inflation of the test statistic. Here, we show by theory, simulation and analysis of data that in the absence of population structure and other technical artefacts, but in the presence of polygenic inheritance, substantial genomic inflation is expected. Its magnitude depends on sample size, heritability, linkage disequilibrium structure and the number of causal variants. Our predictions are consistent with empirical observations on height in independent samples of ~4000 and ~133,000 individuals.
Figures
References
-
- Maher B. Personal genomes: the case of the missing heritability. Nature. 2008;456:18–21. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
