

Fast identification and removal of sequence contamination from genomic and metagenomic datasets
- PMID: 21408061
- PMCID: PMC3052304
- DOI: 10.1371/journal.pone.0017288
Fast identification and removal of sequence contamination from genomic and metagenomic datasets
Abstract
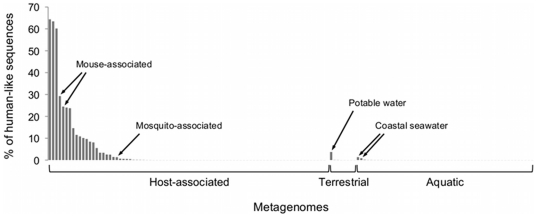
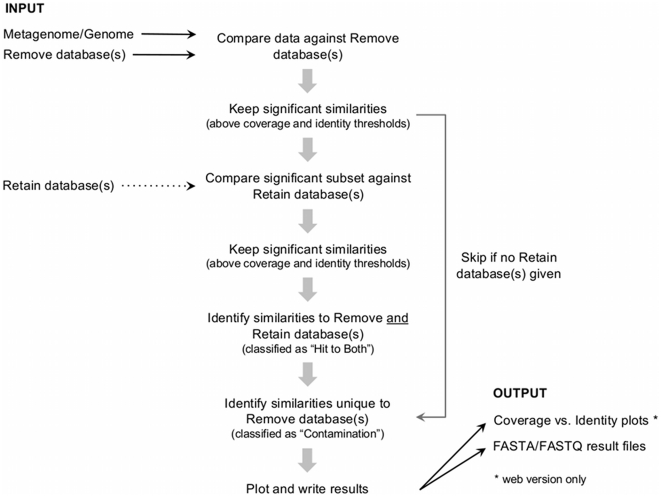
High-throughput sequencing technologies have strongly impacted microbiology, providing a rapid and cost-effective way of generating draft genomes and exploring microbial diversity. However, sequences obtained from impure nucleic acid preparations may contain DNA from sources other than the sample. Those sequence contaminations are a serious concern to the quality of the data used for downstream analysis, causing misassembly of sequence contigs and erroneous conclusions. Therefore, the removal of sequence contaminants is a necessary and required step for all sequencing projects. We developed DeconSeq, a robust framework for the rapid, automated identification and removal of sequence contamination in longer-read datasets (150 bp mean read length). DeconSeq is publicly available as standalone and web-based versions. The results can be exported for subsequent analysis, and the databases used for the web-based version are automatically updated on a regular basis. DeconSeq categorizes possible contamination sequences, eliminates redundant hits with higher similarity to non-contaminant genomes, and provides graphical visualizations of the alignment results and classifications. Using DeconSeq, we conducted an analysis of possible human DNA contamination in 202 previously published microbial and viral metagenomes and found possible contamination in 145 (72%) metagenomes with as high as 64% contaminating sequences. This new framework allows scientists to automatically detect and efficiently remove unwanted sequence contamination from their datasets while eliminating critical limitations of current methods. DeconSeq's web interface is simple and user-friendly. The standalone version allows offline analysis and integration into existing data processing pipelines. DeconSeq's results reveal whether the sequencing experiment has succeeded, whether the correct sample was sequenced, and whether the sample contains any sequence contamination from DNA preparation or host. In addition, the analysis of 202 metagenomes demonstrated significant contamination of the non-human associated metagenomes, suggesting that this method is appropriate for screening all metagenomes. DeconSeq is available at http://deconseq.sourceforge.net/.
Conflict of interest statement
Figures
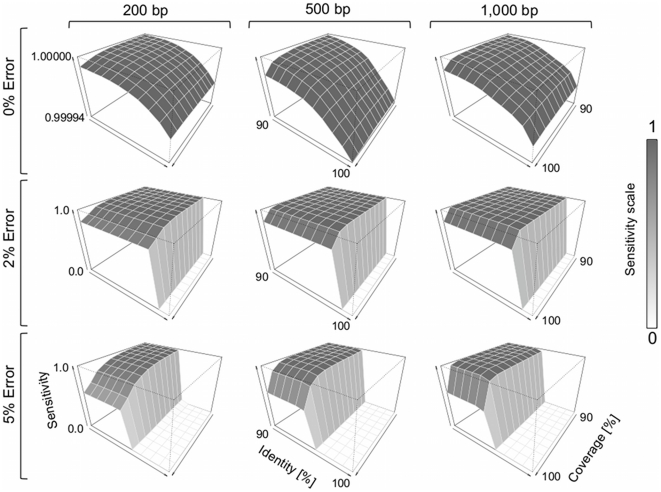
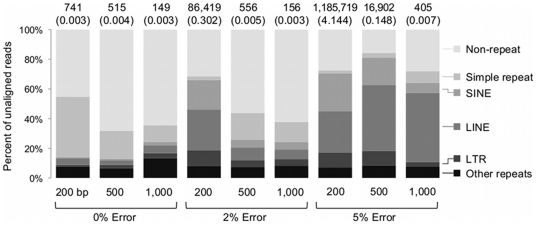
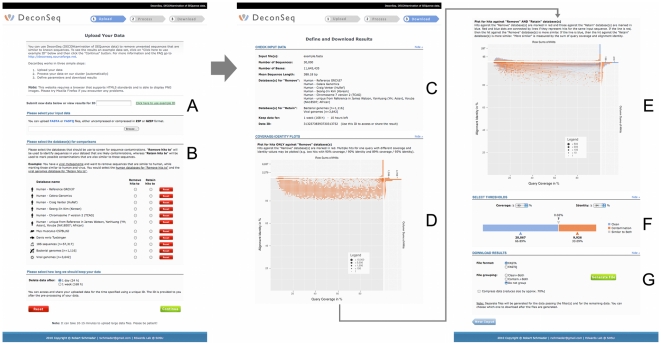
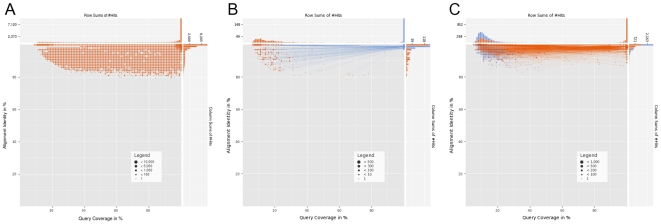
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
