

Mutations in PINK1 and Parkin impair ubiquitination of Mitofusins in human fibroblasts
- PMID: 21408142
- PMCID: PMC3050809
- DOI: 10.1371/journal.pone.0016746
Mutations in PINK1 and Parkin impair ubiquitination of Mitofusins in human fibroblasts
Abstract
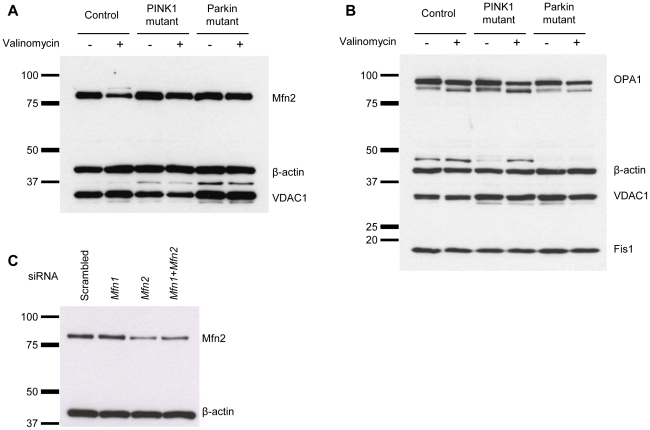
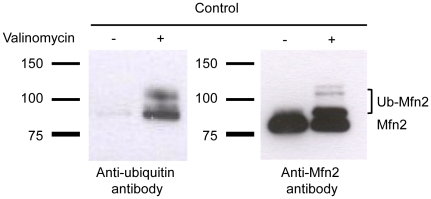
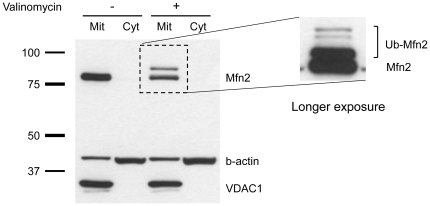
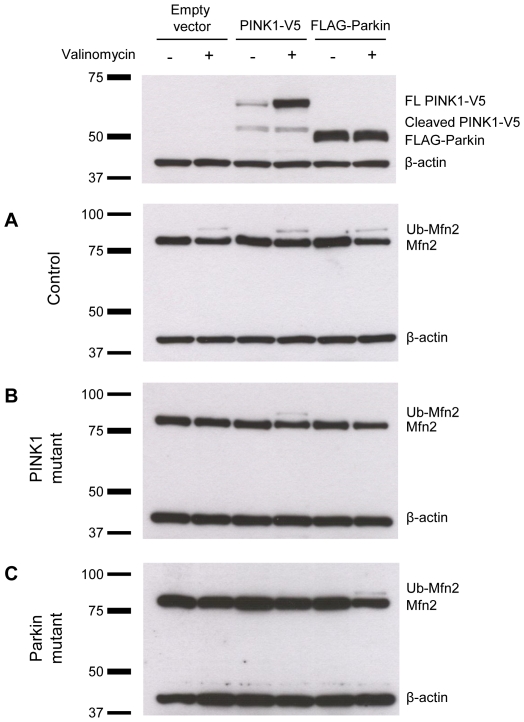
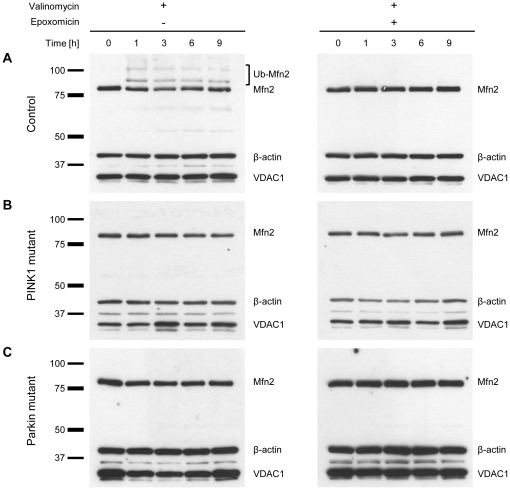
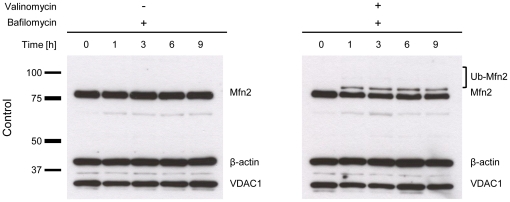
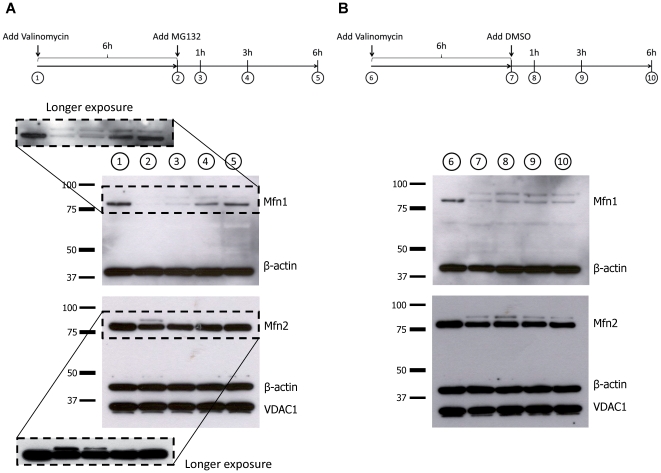
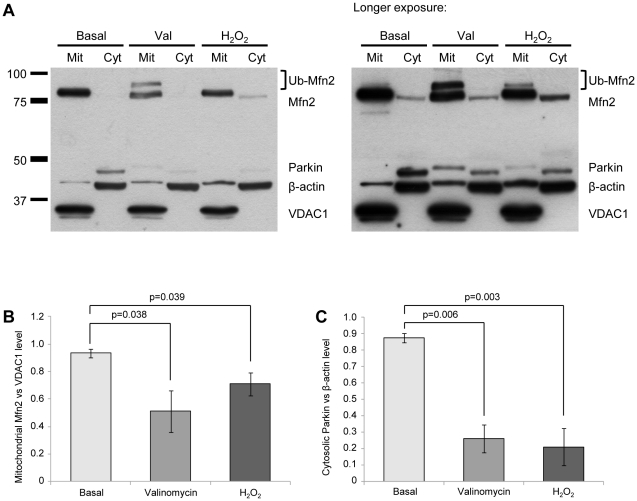
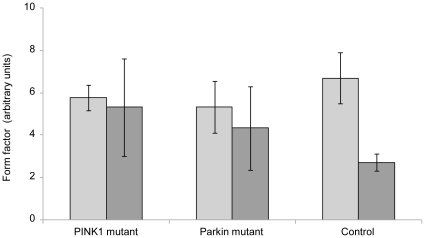
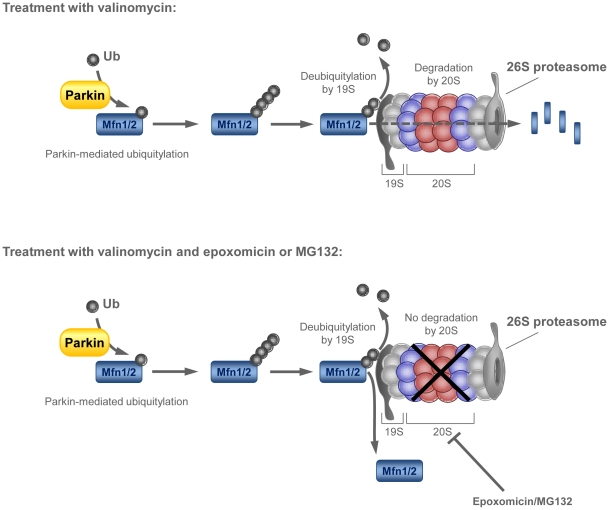
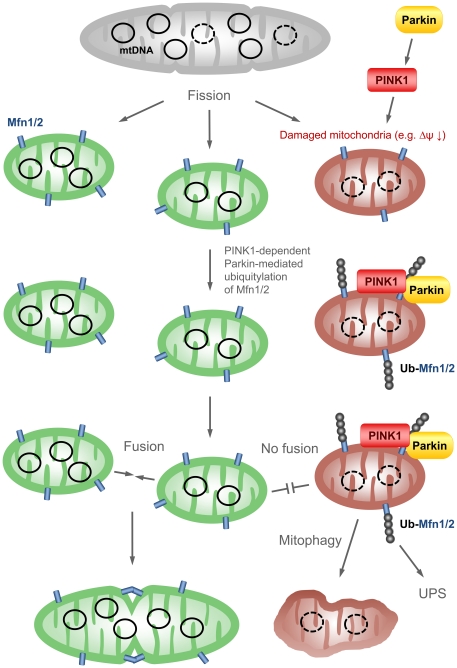
PINK1 and Parkin mutations cause recessive Parkinson's disease (PD). In Drosophila and SH-SY5Y cells, Parkin is recruited by PINK1 to damaged mitochondria, where it ubiquitinates Mitofusins and consequently promotes mitochondrial fission and mitophagy.Here, we investigated the impact of mutations in endogenous PINK1 and Parkin on the ubiquitination of mitochondrial fusion and fission factors and the mitochondrial network structure. Treating control fibroblasts with mitochondrial membrane potential (Δψ) inhibitors or H(2)O(2) resulted in ubiquitination of Mfn1/2 but not of OPA1 or Fis1. Ubiquitination of Mitofusins through the PINK1/Parkin pathway was observed within 1 h of treatment. Upon combined inhibition of Δψ and the ubiquitin proteasome system (UPS), no ubiquitination of Mitofusins was detected. Regarding morphological changes, we observed a trend towards increased mitochondrial branching in PD patient cells upon mitochondrial stress.For the first time in PD patient-derived cells, we demonstrate that mutations in PINK1 and Parkin impair ubiquitination of Mitofusins. In the presence of UPS inhibitors, ubiquitinated Mitofusin is deubiquitinated by the UPS but not degraded, suggesting that the UPS is involved in Mitofusin degradation.
Conflict of interest statement
Figures
References
-
- Klein C, Schlossmacher MG. Parkinson disease, 10 years after its genetic revolution: multiple clues to a complex disorder. Neurology. 2007;69:2093–2104. - PubMed
-
- Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, et al. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279:18614–18622. - PubMed
-
- Flinn L, Mortiboys H, Volkmann K, Koster RW, Ingham PW, et al. Complex I deficiency and dopaminergic neuronal cell loss in parkin-deficient zebrafish (Danio rerio). Brain. 2009;132:1613–1623. - PubMed
-
- Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, et al. Mitochondrial complex I deficiency in Parkinson's disease. Lancet. 1989;1:1269. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
