

The ΔF508 mutation causes CFTR misprocessing and cystic fibrosis-like disease in pigs
- PMID: 21411740
- PMCID: PMC3119077
- DOI: 10.1126/scitranslmed.3001868
The ΔF508 mutation causes CFTR misprocessing and cystic fibrosis-like disease in pigs
Abstract
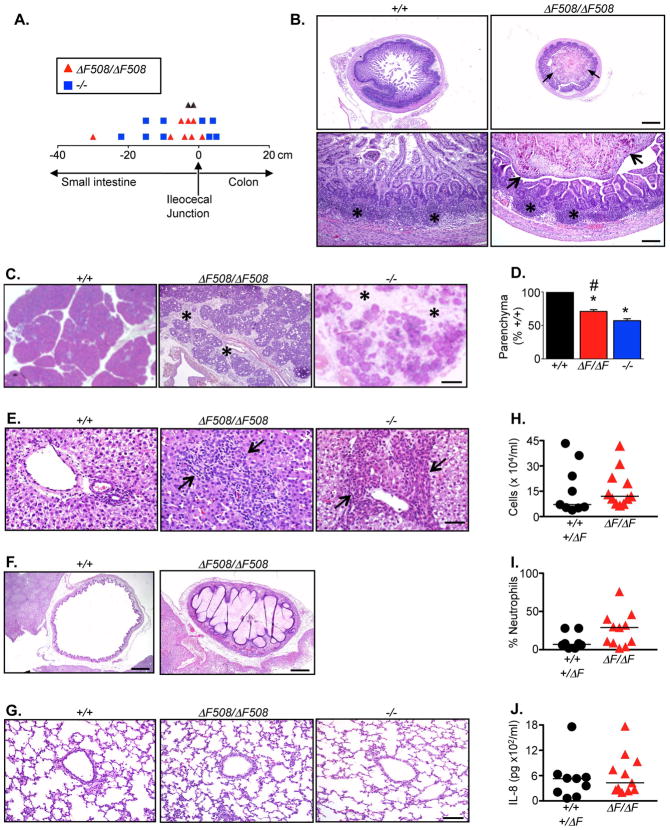
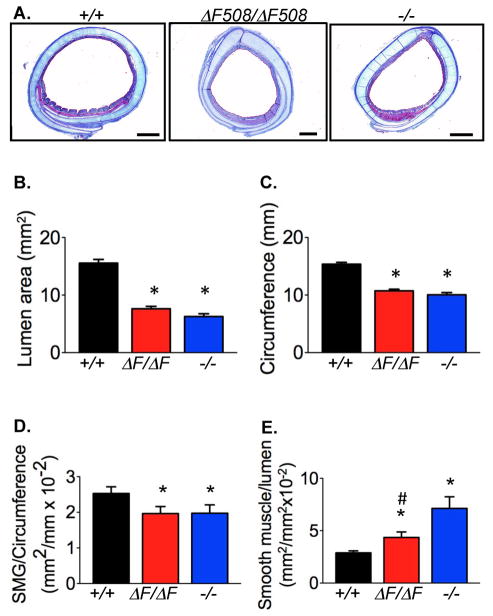
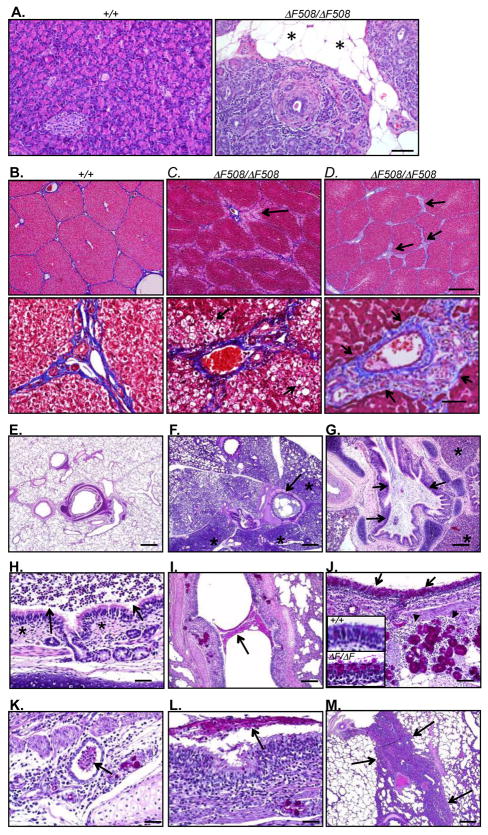
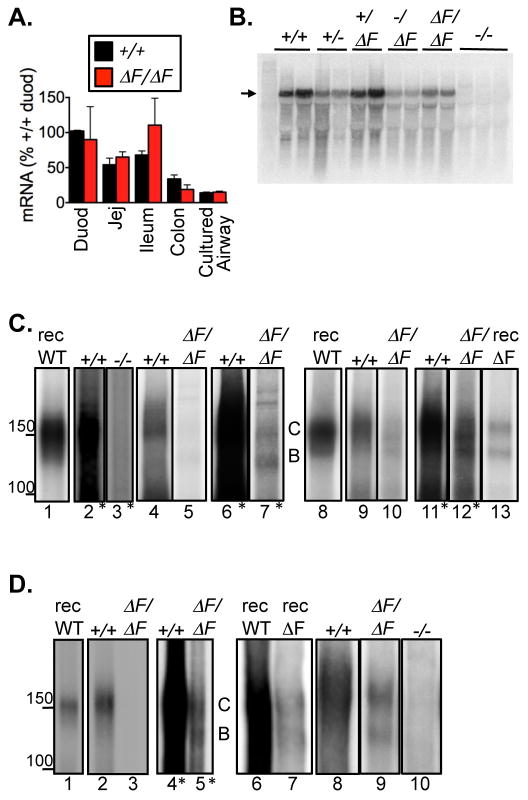
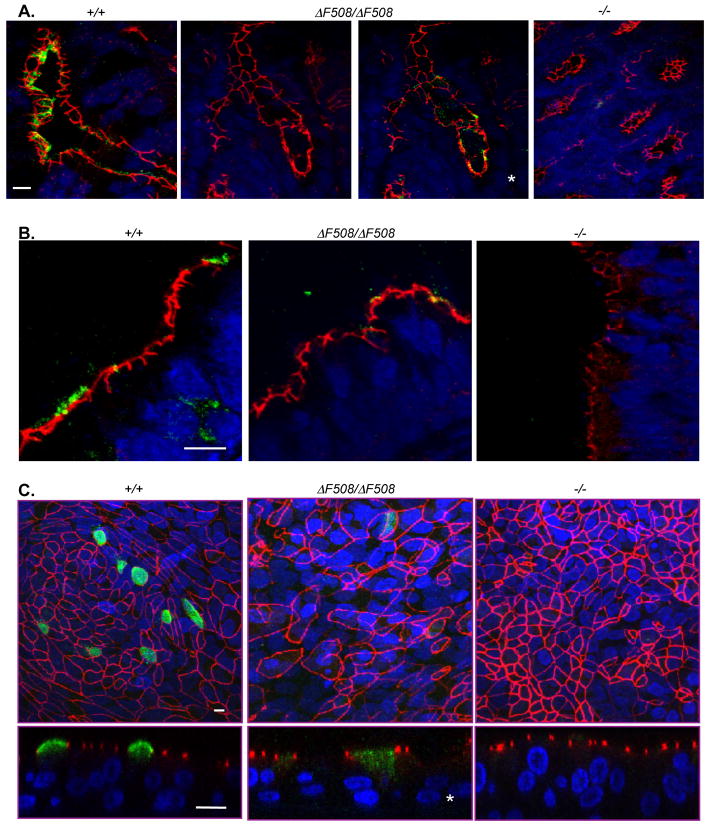
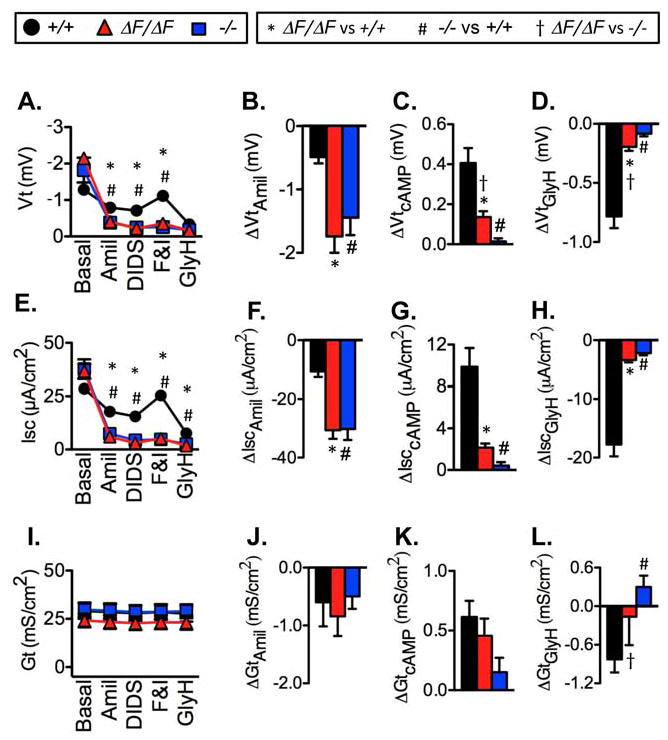
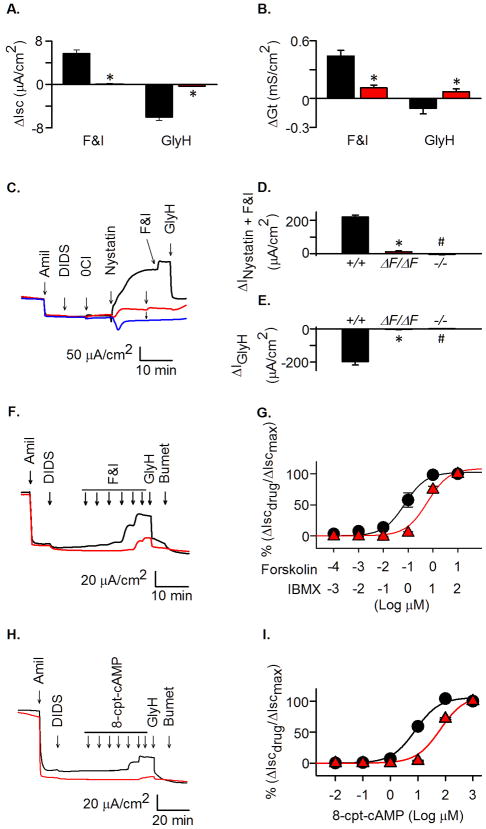
Cystic fibrosis (CF) is an autosomal recessive disease caused by mutations in the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) anion channel. The most common CF-associated mutation is ΔF508, which deletes a phenylalanine in position 508. In vitro studies indicate that the resultant protein, CFTR-ΔF508, is misprocessed, although the in vivo consequences of this mutation remain uncertain. To better understand the effects of the ΔF508 mutation in vivo, we produced CFTR(ΔF508/ΔF508) pigs. Our biochemical, immunocytochemical, and electrophysiological data on CFTR-ΔF508 in newborn pigs paralleled in vitro predictions. They also indicated that CFTR(ΔF508/ΔF508) airway epithelia retain a small residual CFTR conductance, with maximal stimulation producing ~6% of wild-type function. Cyclic adenosine 3',5'-monophosphate (cAMP) agonists were less potent at stimulating current in CFTR(Δ)(F508/)(Δ)(F508) epithelia, suggesting that quantitative tests of maximal anion current may overestimate transport under physiological conditions. Despite residual CFTR function, four older CFTR(ΔF508/ΔF508) pigs developed lung disease similar to human CF. These results suggest that this limited CFTR activity is insufficient to prevent lung or gastrointestinal disease in CF pigs. These data also suggest that studies of recombinant CFTR-ΔF508 misprocessing predict in vivo behavior, which validates its use in biochemical and drug discovery experiments. These findings help elucidate the molecular pathogenesis of the common CF mutation and will guide strategies for developing new therapeutics.
Figures
References
-
- Welsh MJ, Ramsey BW, Accurso F, Cutting GR. Cystic Fibrosis. In: Scriver CR, et al., editors. The Metabolic and Molecular Basis of Inherited Disease. McGraw-Hill; New York: 2001. pp. 5121–5189.
-
- Cheng SH, et al. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. - PubMed
-
- Ward CL, Kopito RR. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J Biol Chem. 1994;269(41):25710–25718. - PubMed
-
- Younger JM, et al. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell. 2006;126(3):571–582. - PubMed
-
- Denning GM, et al. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
