

Carpenter syndrome: extended RAB23 mutation spectrum and analysis of nonsense-mediated mRNA decay
- PMID: 21412941
- PMCID: PMC3429868
- DOI: 10.1002/humu.21457
Carpenter syndrome: extended RAB23 mutation spectrum and analysis of nonsense-mediated mRNA decay
Abstract
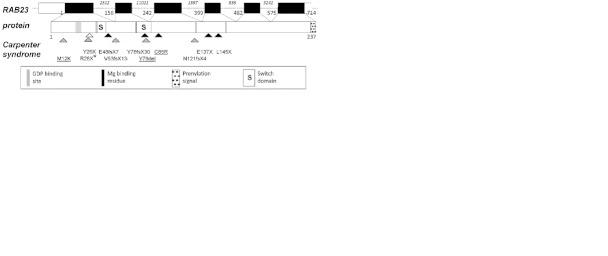
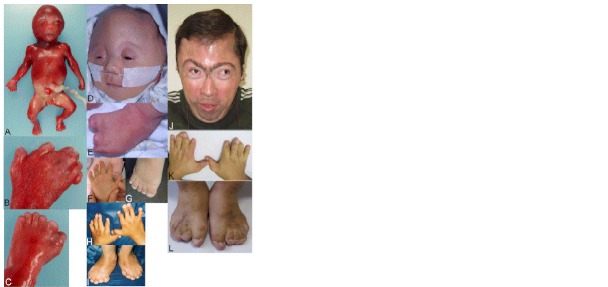
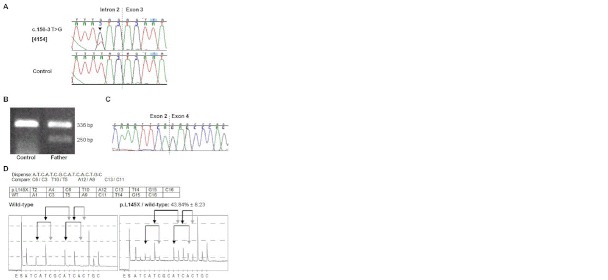
Carpenter syndrome, a rare autosomal recessive disorder characterized by a combination of craniosynostosis, polysyndactyly, obesity, and other congenital malformations, is caused by mutations in RAB23, encoding a member of the Rab-family of small GTPases. In 15 out of 16 families previously reported, the disease was caused by homozygosity for truncating mutations, and currently only a single missense mutation has been identified in a compound heterozygote. Here, we describe a further 8 independent families comprising 10 affected individuals with Carpenter syndrome, who were positive for mutations in RAB23. We report the first homozygous missense mutation and in-frame deletion, highlighting key residues for RAB23 function, as well as the first splice-site mutation. Multi-suture craniosynostosis and polysyndactyly have been present in all patients described to date, and abnormal external genitalia have been universal in boys. High birth weight was not evident in the current group of patients, but further evidence for laterality defects is reported. No genotype-phenotype correlations are apparent. We provide experimental evidence that transcripts encoding truncating mutations are subject to nonsense-mediated decay, and that this plays an important role in the pathogenesis of many RAB23 mutations. These observations refine the phenotypic spectrum of Carpenter syndrome and offer new insights into molecular pathogenesis.
© 2011 Wiley-Liss, Inc.
Figures
References
-
- Alessandri JL, Dagoneau N, Laville JM, Baruteau J, Hébert JC, Cormier-Daire V. RAB23 mutation in a large family from Comoros Islands with Carpenter syndrome. Am J Med Genet A. 2010;152A:982–6. - PubMed
-
- Carpenter G. Two sisters showing malformations of the skull and other congenital abnormalities. Rep Soc Study Dis Child Lond. 1901;1:110–118.
-
- Hall GW, Thein S. Nonsense codon mutations in the terminal exon of the β-globin gene are not associated with a reduction in β-mRNA accumulation - A mechanism for the phenotype of dominant β-thalassemia. Blood. 1994;83:2031–2037. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
