

ATAQS: A computational software tool for high throughput transition optimization and validation for selected reaction monitoring mass spectrometry
- PMID: 21414234
- PMCID: PMC3213215
- DOI: 10.1186/1471-2105-12-78
ATAQS: A computational software tool for high throughput transition optimization and validation for selected reaction monitoring mass spectrometry
Abstract
Background: Since its inception, proteomics has essentially operated in a discovery mode with the goal of identifying and quantifying the maximal number of proteins in a sample. Increasingly, proteomic measurements are also supporting hypothesis-driven studies, in which a predetermined set of proteins is consistently detected and quantified in multiple samples. Selected reaction monitoring (SRM) is a targeted mass spectrometric technique that supports the detection and quantification of specific proteins in complex samples at high sensitivity and reproducibility. Here, we describe ATAQS, an integrated software platform that supports all stages of targeted, SRM-based proteomics experiments including target selection, transition optimization and post acquisition data analysis. This software will significantly facilitate the use of targeted proteomic techniques and contribute to the generation of highly sensitive, reproducible and complete datasets that are particularly critical for the discovery and validation of targets in hypothesis-driven studies in systems biology.
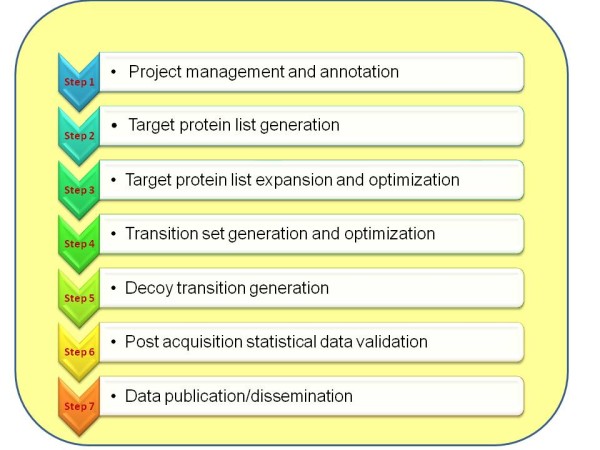
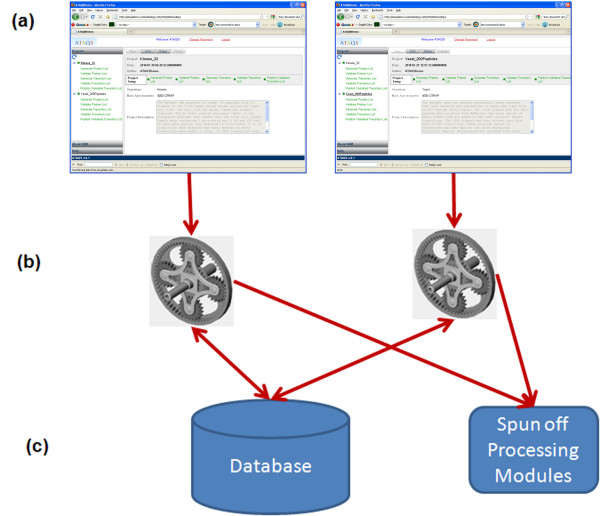
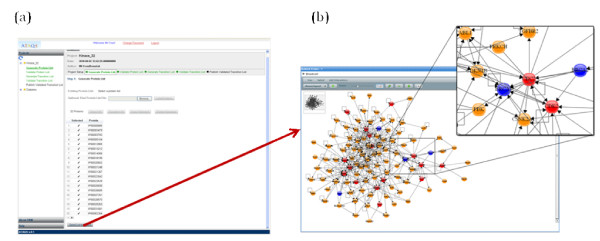
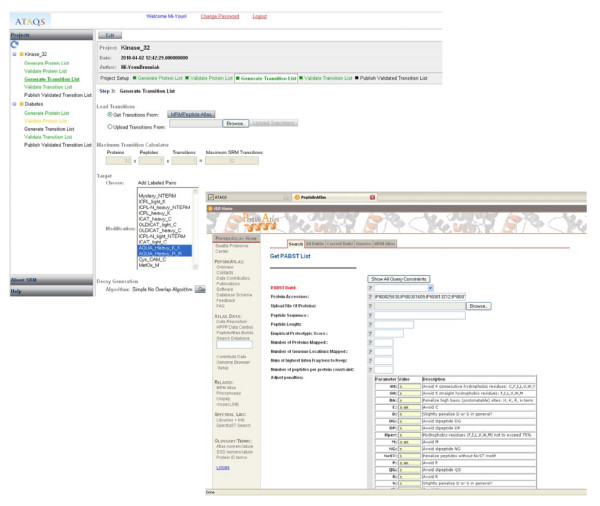
Result: We introduce a new open source software pipeline, ATAQS (Automated and Targeted Analysis with Quantitative SRM), which consists of a number of modules that collectively support the SRM assay development workflow for targeted proteomic experiments (project management and generation of protein, peptide and transitions and the validation of peptide detection by SRM). ATAQS provides a flexible pipeline for end-users by allowing the workflow to start or end at any point of the pipeline, and for computational biologists, by enabling the easy extension of java algorithm classes for their own algorithm plug-in or connection via an external web site.This integrated system supports all steps in a SRM-based experiment and provides a user-friendly GUI that can be run by any operating system that allows the installation of the Mozilla Firefox web browser.
Conclusions: Targeted proteomics via SRM is a powerful new technique that enables the reproducible and accurate identification and quantification of sets of proteins of interest. ATAQS is the first open-source software that supports all steps of the targeted proteomics workflow. ATAQS also provides software API (Application Program Interface) documentation that enables the addition of new algorithms to each of the workflow steps. The software, installation guide and sample dataset can be found in http://tools.proteomecenter.org/ATAQS/ATAQS.html.
Figures
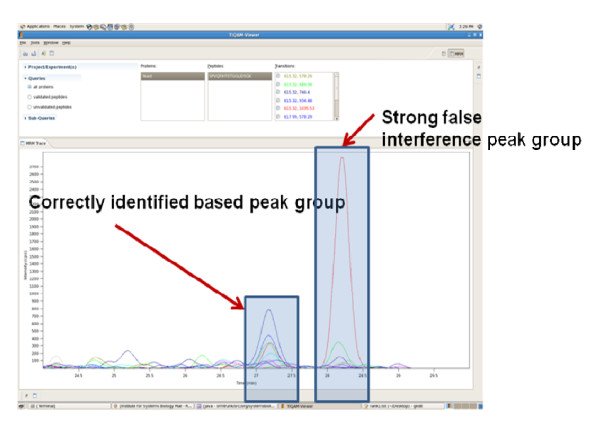
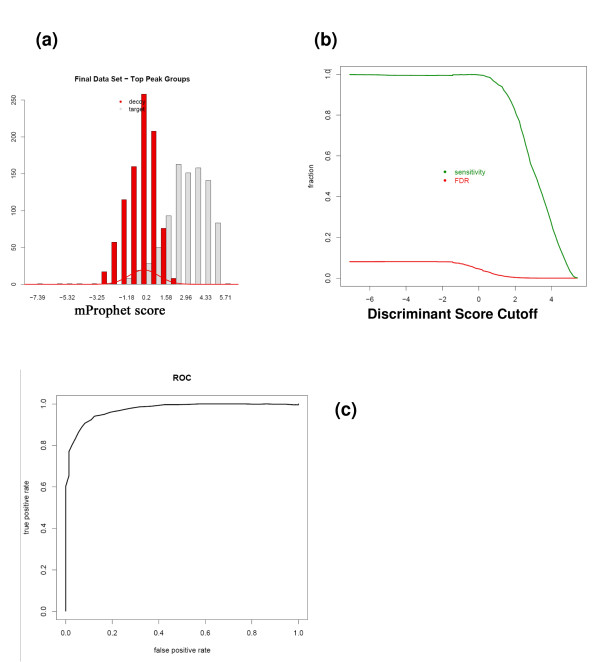
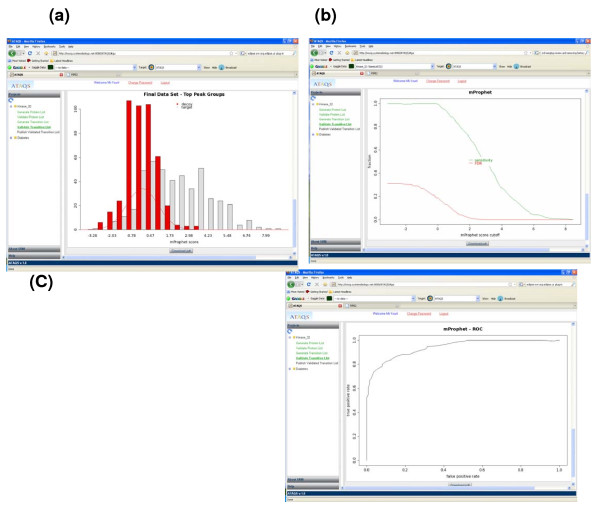
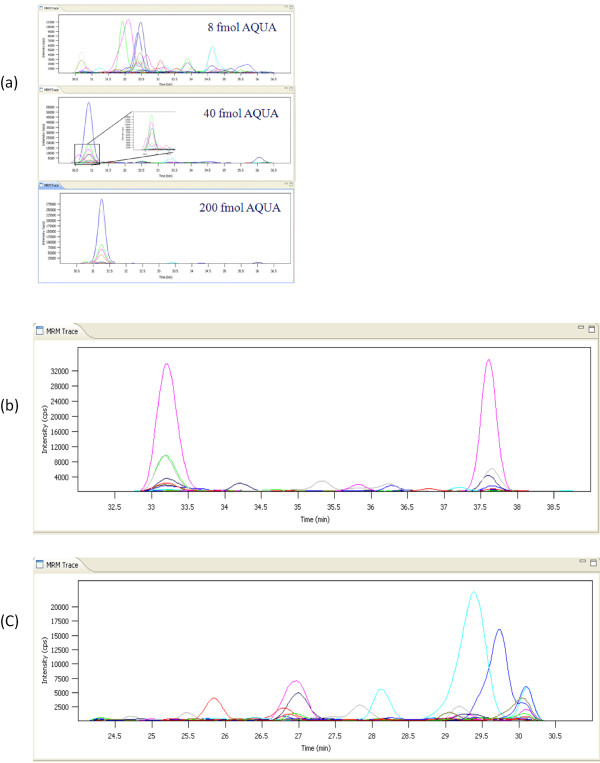
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
