

Spectrum of mutations in Gitelman syndrome
- PMID: 21415153
- PMCID: PMC3065225
- DOI: 10.1681/ASN.2010090907
Spectrum of mutations in Gitelman syndrome
Abstract
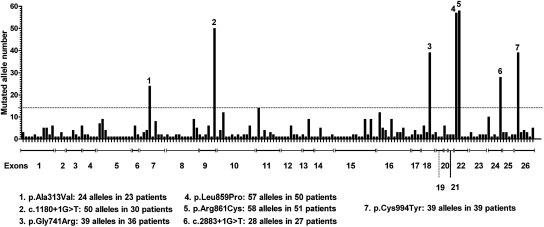
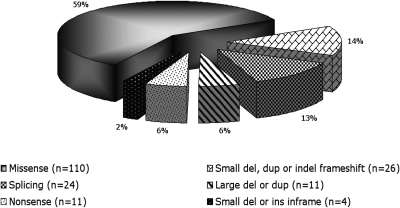
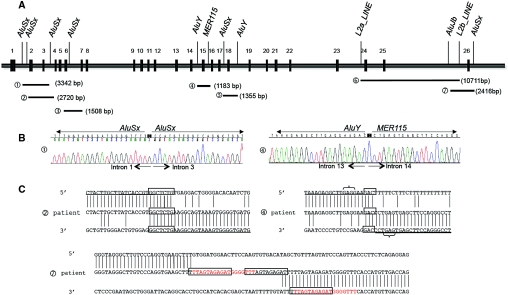
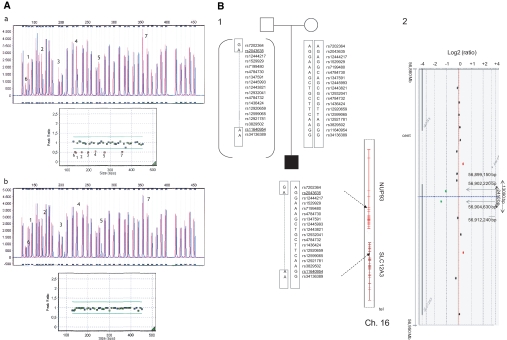
Gitelman's syndrome (GS) is a rare, autosomal recessive, salt-losing tubulopathy caused by mutations in the SLC12A3 gene, which encodes the thiazide-sensitive NaCl cotransporter (NCC). Because 18 to 40% of suspected GS patients carry only one SLC12A3 mutant allele, large genomic rearrangements may account for unidentified mutations. Here, we directly sequenced genomic DNA from a large cohort of 448 unrelated patients suspected of having GS. We found 172 distinct mutations, of which 100 were unreported previously. In 315 patients (70%), we identified two mutations; in 81 patients (18%), we identified one; and in 52 patients (12%), we did not detect a mutation. In 88 patients, we performed a search for large rearrangements by multiplex ligation-dependent probe amplification (MLPA) and found nine deletions and two duplications in 24 of the 51 heterozygous patients. A second technique confirmed each rearrangement. Based on the breakpoints of seven deletions, nonallelic homologous recombination by Alu sequences and nonhomologous end-joining probably favor these intragenic deletions. In summary, missense mutations account for approximately 59% of the mutations in Gitelman's syndrome, and there is a predisposition to large rearrangements (6% of our cases) caused by the presence of repeated sequences within the SLC12A3 gene.
Copyright © 2011 by the American Society of Nephrology
Figures
References
-
- Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, Vaara I, Iwata F, Cushner HM, Koolen M, Gainza FJ, Gitleman HJ, Lifton RP: Gitelman's variant of Bartter's syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet 12: 24–30, 1996 - PubMed
-
- Mastroianni N, De Fusco M, Zollo M, Arrigo G, Zuffardi O, Bettinelli A, Ballabio A, Casari G: Molecular cloning, expression pattern, and chromosomal localization of the human Na-Cl thiazide-sensitive cotransporter (SLC12A3). Genomics 35: 486–493, 1996 - PubMed
-
- Melander O, Orho-Melander M, Bengtsson K, Lindblad U, Rastam L, Groop L, Hulthen UL: Genetic variants of thiazide-sensitive NaCl-cotransporter in Gitelman's syndrome and primary hypertension. Hypertension, 36: 389–394, 2000 - PubMed
-
- Lemmink HH, Knoers NV, Karolyi L, van Dijk H, Niaudet P, Antignac C, Guay-Woodford LM, Goodyer PR, Carel JC, Hermes A, Seyberth HW, Monnens LA, van den Heuvel LP: Novel mutations in the thiazide-sensitive NaCl cotransporter gene in patients with Gitelman syndrome with predominant localization to the C-terminal domain. Kidney Int 54: 720–730, 1998 - PubMed
-
- Riveira-Munoz E, Chang Q, Godefroid N, Hoenderop JG, Bindels RJ, Dahan K, Devuyst O: Transcriptional and functional analyses of SLC12A3 mutations: New clues for the pathogenesis of Gitelman syndrome. J Am Soc Nephrol 18: 1271–1283, 2007 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
