

Proliferative glomerulonephritis secondary to dysfunction of the alternative pathway of complement
- PMID: 21415311
- PMCID: PMC3087765
- DOI: 10.2215/CJN.07110810
Proliferative glomerulonephritis secondary to dysfunction of the alternative pathway of complement
Abstract
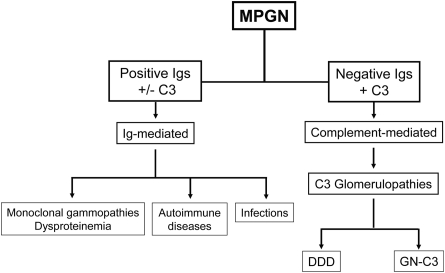
Background and objectives: dense deposit disease (DDD) is the prototypical membranoproliferative glomerulonephritis (MPGN), in which fluid-phase dysregulation of the alternative pathway (AP) of complement results in the accumulation of complement debris in the glomeruli, often producing an MPGN pattern of injury in the absence of immune complexes. A recently described entity referred to as GN with C3 deposition (GN-C3) bears many similarities to DDD. The purpose of this study was to evaluate AP function in cases of GN-C3.
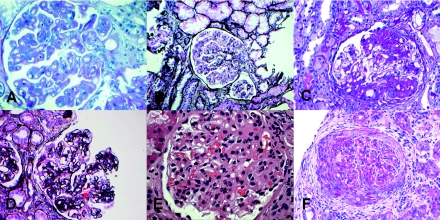
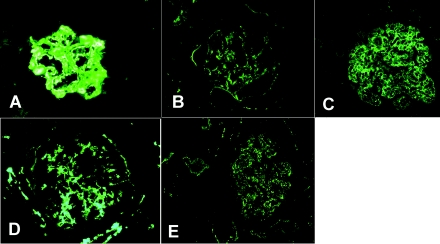
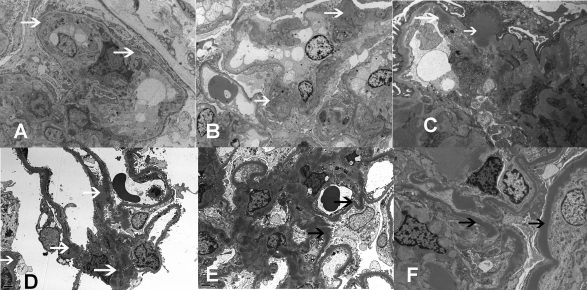
Design, setting, participants, & measurements: Five recent cases of MPGN with extensive C3 deposition were studied. Renal biopsy in one case exhibited the classic findings of DDD. Three cases showed GN-C3 in the absence of significant Ig deposition; however, the classic hallmark of DDD-dense deposits along the glomerular basement membranes and mesangium-was absent. The remaining case exhibited features of both DDD and GN-C3.
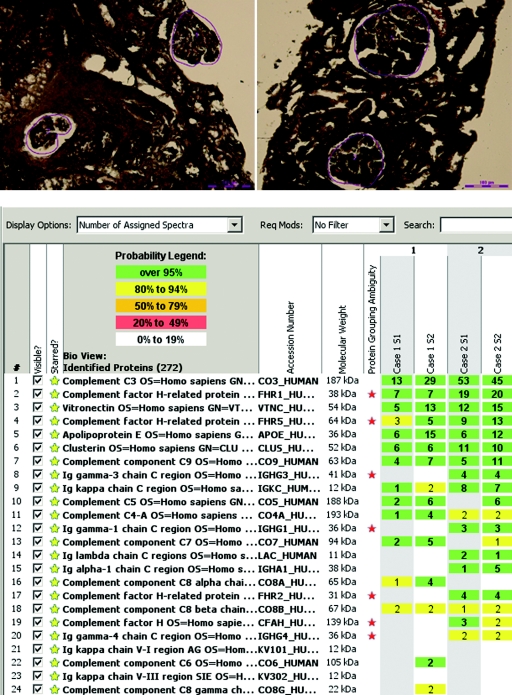
Results: Evidence of AP activation was demonstrable in all cases and included increased levels of soluble membrane attack complex (all cases), positive AP functional assays (four cases), and a positive hemolytic assay (one case). Autoantibodies were found to C3 convertase (two cases) and to factor H (one case). Factor H mutation screening identified the H402 allele (all cases) and a c.C2867T p.T956M missence mutation (one case). Laser microdissection and mass spectrometry of glomeruli of GN-C3 (two cases) showed a proteomic profile very similar to DDD.
Conclusions: These studies implicate AP dysregulation in a spectrum of rare renal diseases that includes GN-C3 and DDD.
Copyright © 2011 by the American Society of Nephrology
Figures
References
-
- Rennke H: Secondary membranoproliferative glomerulonephritis. Kidney Int 47: 643–656, 1995 - PubMed
-
- Alpers CE, Smith KD: Cryoglobulinemia and renal disease. Curr Opin Nephrol Hypertens 17: 243–249, 2008 - PubMed
-
- Appel GB, Cook HT, Hageman G, Jennette JC, Kashgarian M, Kirschfink M, Lambris JD, Lanning L, Lutz HU, Meri S, Rose NR, Salant DJ, Sethi S, Smith RJ, Smoyer W, Tully HF, Tully SP, Walker P, Welsh M, Wurzner R, Zipfel PF: Membranoproliferative glomerulonephritis type II (dense deposit disease): An update. J Am Soc Nephrol 16: 1392–1403, 2005 - PubMed
-
- Smith RJH, Alexander J, Barlow PN, Botto M, Cassavant TL, Cook HT, de Córdoba SR, Hageman GS, Jokiranta TS, Kimberling WJ, Lambris JD, Lanning LD, Levidiotis V, Licht C, Lutz HU, Meri S, Pickering MC, Quigg RJ, Rops AL, Salant DJ, Sethi S, Thurman JM, Tully HF, Tully SP, van der Vlag J, Walker PD, Würzner R, Zipfel PF; Dense Deposit Disease Focus Group: New approaches to the treatment of dense deposit disease. J Am Soc Nephrol 18: 2447–2456, 2007 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
