

Sepsis-associated disseminated intravascular coagulation and thromboembolic disease
- PMID: 21415977
- PMCID: PMC3033145
- DOI: 10.4084/MJHID.2010.024
Sepsis-associated disseminated intravascular coagulation and thromboembolic disease
Abstract
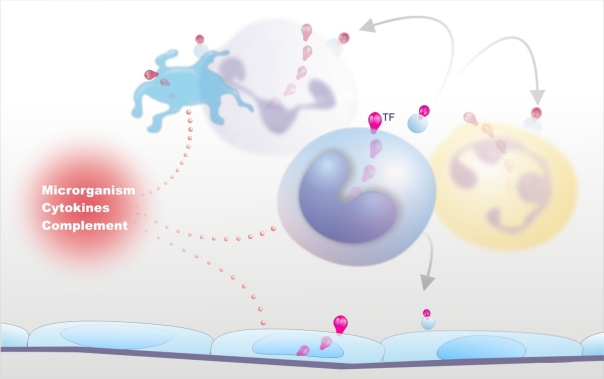
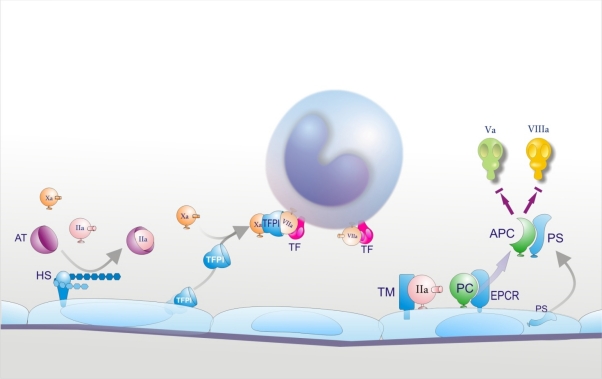
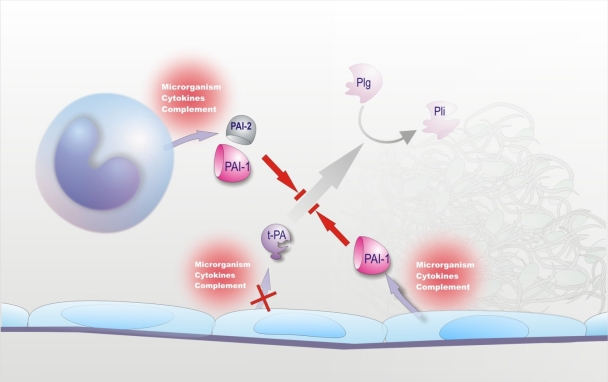
Sepsis is almost invariably associated with haemostatic abnormalities ranging from subclinical activation of blood coagulation (hypercoagulability), which may contribute to localized venous thromboembolism, to acute disseminated intravascular coagulation (DIC), characterized by massive thrombin formation and widespread microvascular thrombosis, partly responsible of the multiple organ dysfunction syndrome (MODS), and subsequent consumption of platelets and coagulation proteins causing, in most severe cases, bleeding manifestations. There is general agreement that the key event underlying this life-threatening sepsis complication is the overwhelming inflammatory host response to the infectious agent leading to the overexpression of inflammatory mediators. Mechanistically, the latter, together with the micro-organism and its derivatives, causes DIC by 1) up-regulation of procoagulant molecules, primarily tissue factor (TF), which is produced mainly by stimulated monocytes-macrophages and by specific cells in target tissues; 2) impairment of physiological anticoagulant pathways (antithrombin, protein C pathway, tissue factor pathway inhibitor), which is orchestrated mainly by dysfunctional endothelial cells (ECs); and 3) suppression of fibrinolysis due to increased plasminogen activator inhibitor-1 (PAI-1) by ECs and likely also to thrombin-mediated activation of thrombin-activatable fibrinolysis inhibitor (TAFI). Notably, clotting enzymes non only lead to microvascular thrombosis but can also elicit cellular responses that amplify the inflammatory reactions. Inflammatory mediators can also cause, directly or indirectly, cell apoptosis or necrosis and recent evidence indicates that products released from dead cells, such as nuclear proteins (particularly extracellular histones), are able to propagate further inflammation, coagulation, cell death and MODS. These insights into the pathogenetic mechanisms of DIC and MODS may have important implications for the development of new therapeutic agents that could be potentially useful particularly for the management of severe sepsis.
Figures
References
-
- Levi M, Schultz M, van der Poll T. Disseminated intravascular coagulation in infectious disease. Semin Thromb Hemost. 2010;36:367–77. - PubMed
-
- van Gorp EC, Suharti C, ten Cate H, Dolmans WM, van der Meer JW, ten Cate JW, Brandjes DP. Review: infectious diseases and coagulation disorders. J Infect Dis. 1999;180:176–86. - PubMed
-
- Smeeth L, Cook C, Thomas S, Hall AJ, Hubbard R, Vallance P. Risk of deep vein thrombosis and pulmonary embolism after acute infection in a community setting. Lancet. 2006;367:1075–9. - PubMed
-
- Alikhan R, Spyropoulos AC. Epidemiology of venous thromboembolism in cardiorespiratory and infectious disease. Am J Med. 2008;121:935–42. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous