

Familial prion disease with Alzheimer disease-like tau pathology and clinical phenotype
- PMID: 21416485
- PMCID: PMC3114566
- DOI: 10.1002/ana.22264
Familial prion disease with Alzheimer disease-like tau pathology and clinical phenotype
Abstract
Objective: To describe the Alzheimer disease (AD)-like clinical and pathological features, including marked neurofibrillary tangle (NFT) pathology, of a familial prion disease due to a rare nonsense mutation of the prion gene (PRNP).
Methods: Longitudinal clinical assessments were available for the proband and her mother. After death, both underwent neuropathological evaluation. PRNP was sequenced after failure to find immunopositive Aβ deposits in the proband and the documentation of prion protein (PrP) immunopositive pathology.
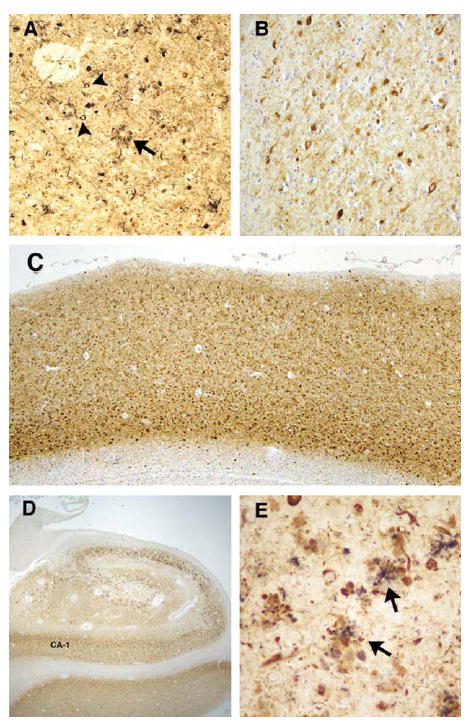
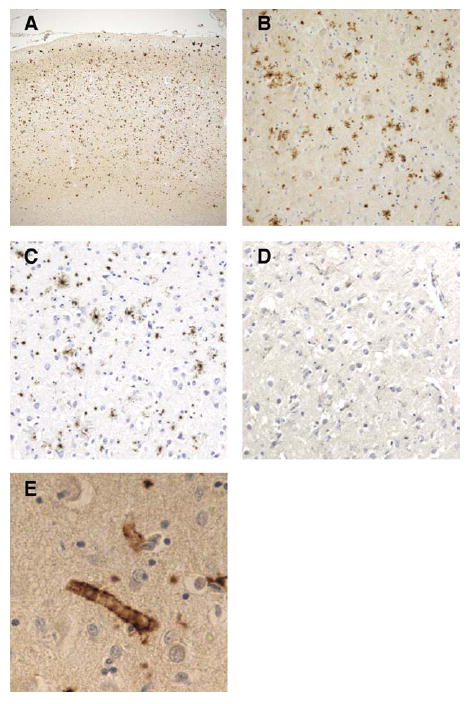
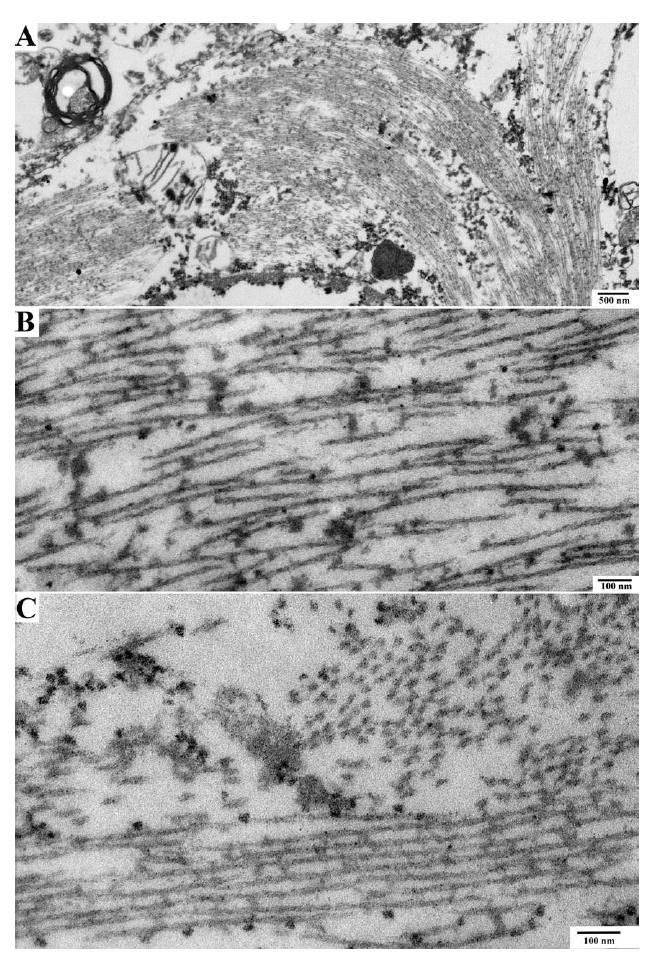
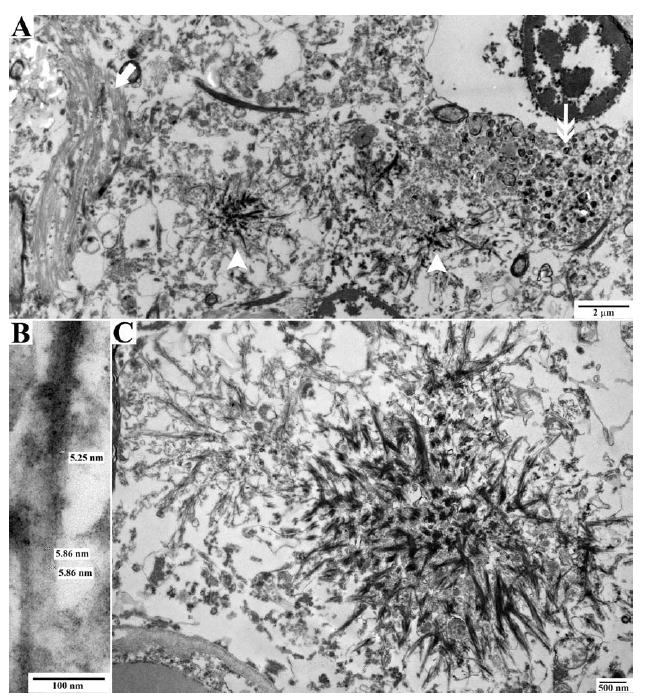
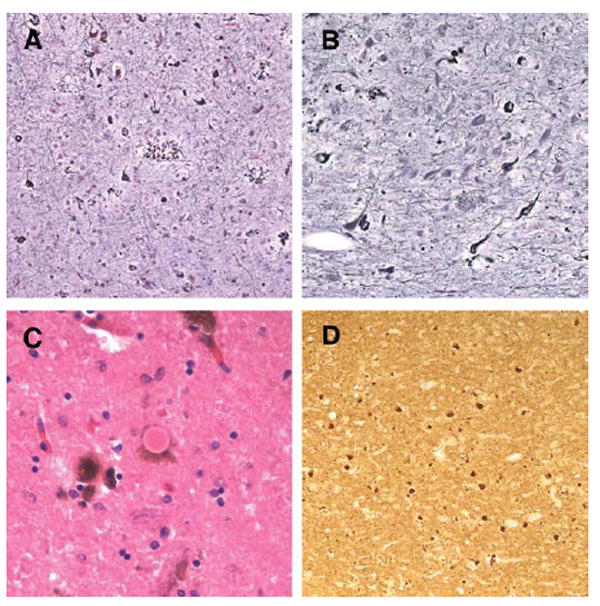
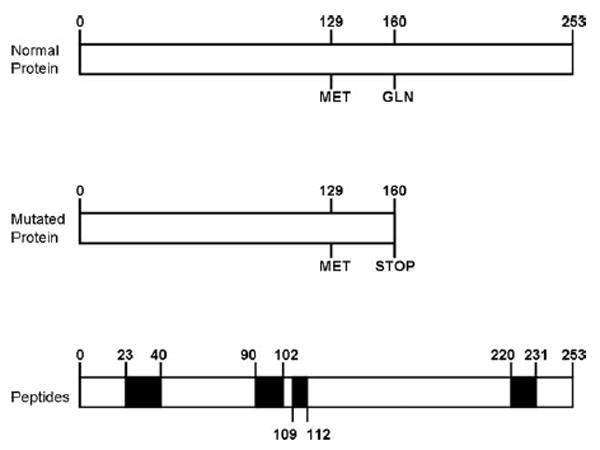
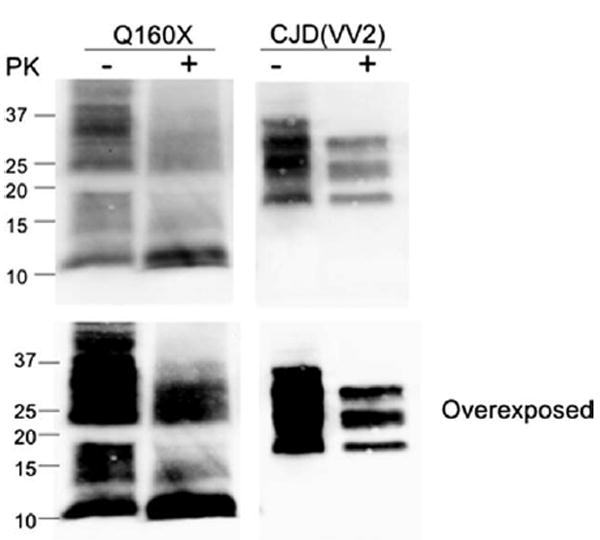
Results: The proband presented at age 42 years with a 3-year history of progressive short-term memory impairment and depression. Neuropsychological testing found impaired memory performance, with relatively preserved attention and construction. She was diagnosed with AD and died at age 47 years. Neuropathologic evaluation revealed extensive limbic and neocortical NFT formation and neuritic plaques consistent with a Braak stage of VI. The NFTs were immunopositive, with multiple tau antibodies, and electron microscopy revealed paired helical filaments. However, the neuritic plaques were immunonegative for Aβ, whereas immunostaining for PrP was positive. The mother of the proband had a similar presentation, including depression, and had been diagnosed clinically and pathologically as AD. Reevaluation of her brain tissue confirmed similar tau and PrP immunostaining findings. Genetic analysis revealed that both the proband and her mother had a rare PRNP mutation (Q160X) that resulted in the production of truncated PrP.
Interpretation: We suggest that PRNP mutations that result in a truncation of PrP lead to a prolonged clinical course consistent with a clinical diagnosis of AD and severe AD-like NFTs.
Copyright © 2010 American Neurological Association.
Conflict of interest statement
Figures
References
-
- Ironside JW, Ghetti B, Head MW. Prions diseases. In: Love S, Louis DN, Ellison DW, editors. Greenfield’s neuropathology. London, UK: Hodder Arnold; 2008. pp. 1197–1274.
-
- Kong Q, Suresicz WK, Petersen RB, et al. Inherited prion diseases. In: Prusiner SB, editor. Prion biology and diseases. 2. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2004. pp. 673–775.
-
- Piccardo P, Ghetti B, Dickson DW, et al. Gerstmann-Straussler-Scheinker disease (PRNP P102L): amyloid deposits are best recognized by antibodies directed to epitopes in PrP region 90-165. J Neuropathol Exp Neurol. 1995;54:790–801. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
