

Prions
- PMID: 21421910
- PMCID: PMC3003464
- DOI: 10.1101/cshperspect.a006833
Prions
Abstract
The discovery of infectious proteins, denoted prions, was unexpected. After much debate over the chemical basis of heredity, resolution of this issue began with the discovery that DNA, not protein, from pneumococcus was capable of genetically transforming bacteria (Avery et al. 1944). Four decades later, the discovery that a protein could mimic viral and bacterial pathogens with respect to the transmission of some nervous system diseases (Prusiner 1982) met with great resistance. Overwhelming evidence now shows that Creutzfeldt-Jakob disease (CJD) and related disorders are caused by prions. The prion diseases are characterized by neurodegeneration and lethality. In mammals, prions reproduce by recruiting the normal, cellular isoform of the prion protein (PrP(C)) and stimulating its conversion into the disease-causing isoform (PrP(Sc)). PrP(C) and PrP(Sc) have distinct conformations: PrP(C) is rich in α-helical content and has little β-sheet structure, whereas PrP(Sc) has less α-helical content and is rich in β-sheet structure (Pan et al. 1993). The conformational conversion of PrP(C) to PrP(Sc) is the fundamental event underlying prion diseases. In this article, we provide an introduction to prions and the diseases they cause.
Figures
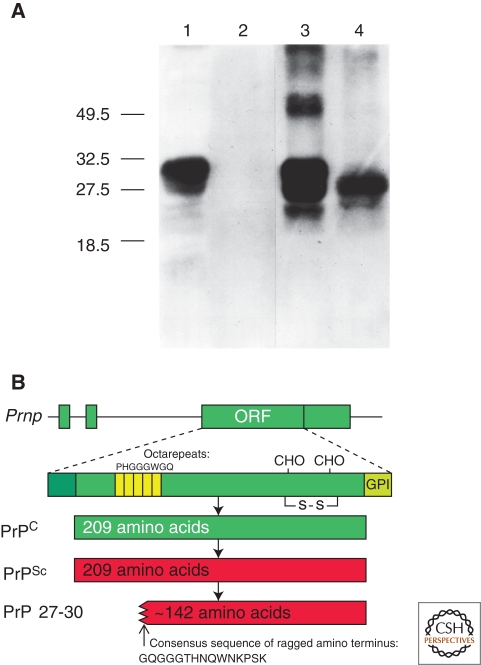
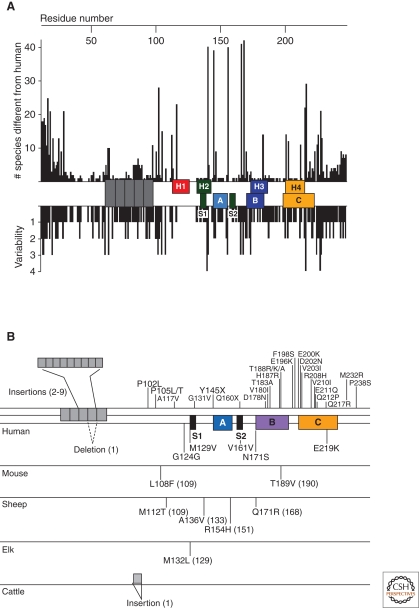
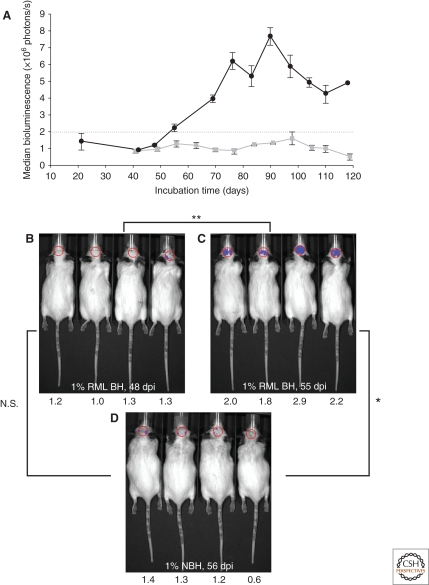
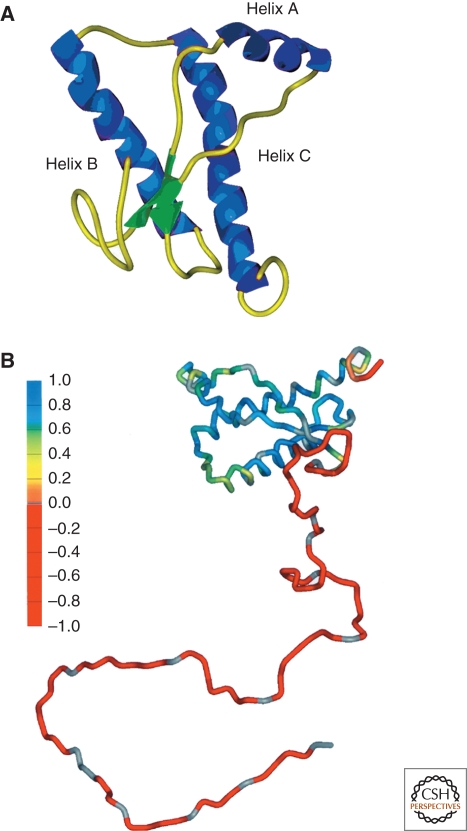
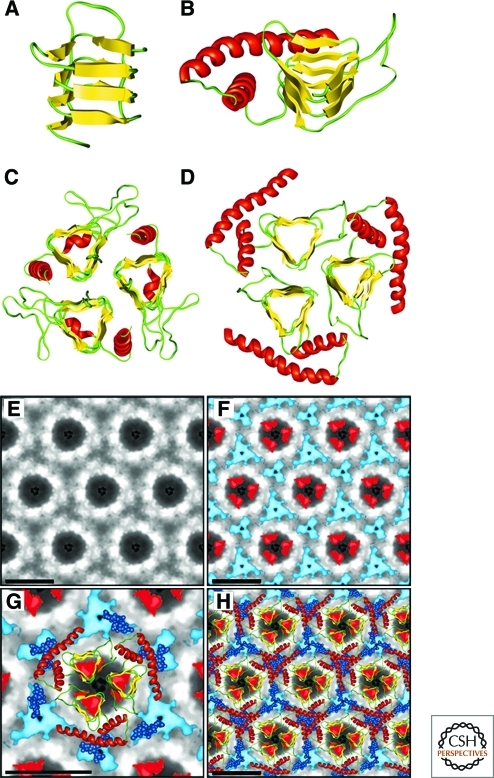
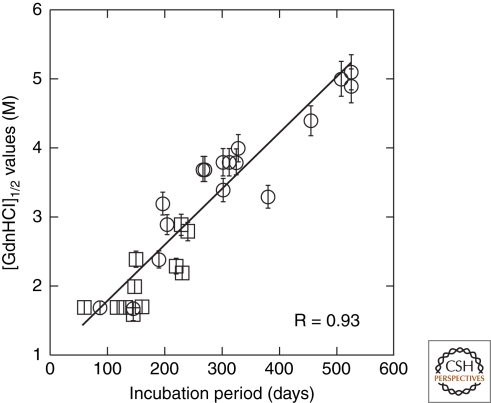
References
-
- Alpers MP 1968. Kuru: implications of its transmissibility for the interpretation of its changing epidemiological pattern. In The central nervous system: some experimental models of neurological diseases. (ed. Bailey OT, Smith DE), pp. 234–251 Williams and Wilkins Company, Baltimore
-
- Anderson RM, Donnelly CA, Ferguson NM, Woolhouse MEJ, Watt CJ, Udy HJ, MaWhinney S, Dunstan SP, Southwood TRE, Wilesmith JW, et al.1996. Transmission dynamics and epidemiology of BSE in British cattle. Nature 382: 779–788 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous