

Emergent properties of proteostasis in managing cystic fibrosis
- PMID: 21421917
- PMCID: PMC3039536
- DOI: 10.1101/cshperspect.a004499
Emergent properties of proteostasis in managing cystic fibrosis
Abstract
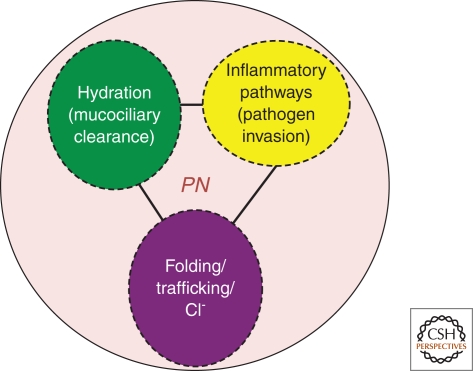
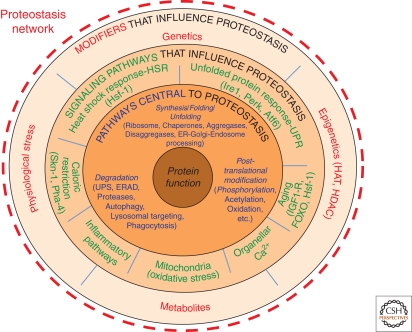
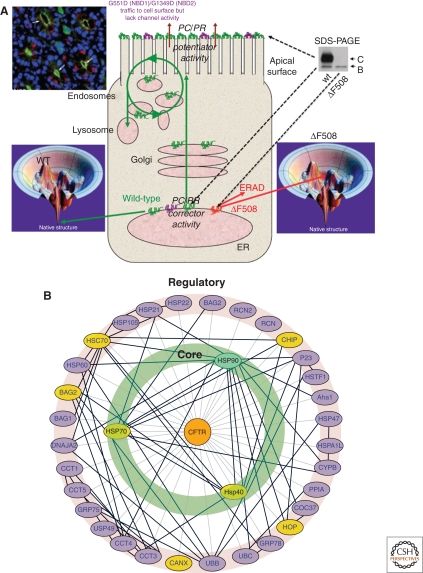
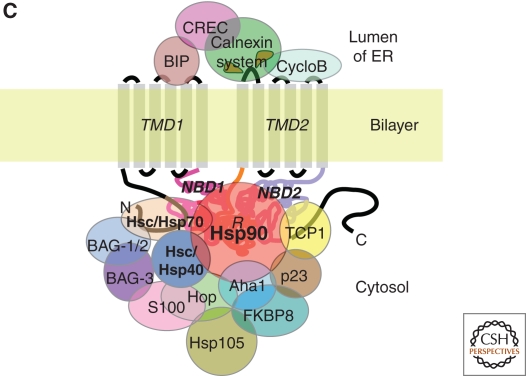
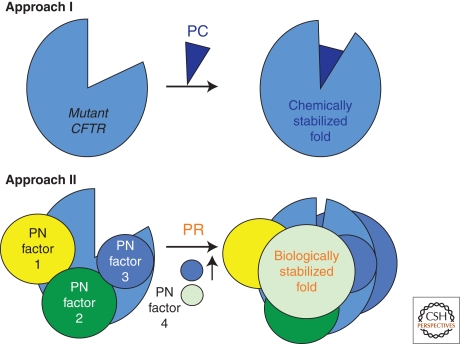
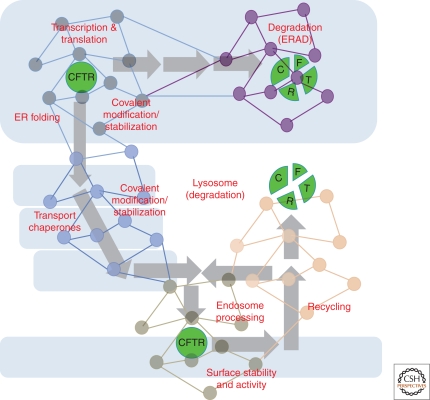
Cystic fibrosis (CF) is a consequence of defective recognition of the multimembrane spanning protein cystic fibrosis conductance transmembrane regulator (CFTR) by the protein homeostasis or proteostasis network (PN) (Hutt and Balch (2010). Like many variant proteins triggering misfolding diseases, mutant CFTR has a complex folding and membrane trafficking itinerary that is managed by the PN to maintain proteome balance and this balance is disrupted in human disease. The biological pathways dictating the folding and function of CFTR in health and disease are being studied by numerous investigators, providing a unique opportunity to begin to understand and therapeutically address the role of the PN in disease onset, and its progression during aging. We discuss the general concept that therapeutic management of the emergent properties of the PN to control the energetics of CFTR folding biology may provide significant clinical benefit.
Figures
References
-
- Amaral MD 2004. CFTR and chaperones: processing and degradation. J Mol Neurosci 23: 41–48 - PubMed
-
- Amaral MD, Kunzelmann K 2007. Molecular targeting of CFTR as a therapeutic approach to cystic fibrosis. Trends Pharmacol Sci 28: 334–341 - PubMed
-
- Balch WE, Morimoto RI, Dillin A, Kelly JW 2008. Adapting proteostasis for disease intervention. Science 319: 916–919 - PubMed
-
- Becq F 2010. Cystic fibrosis transmembrane conductance regulator modulators for personalized drug treatment of cystic fibrosis: progress to date. Drugs 70: 241–259 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical