

HMMerThread: detecting remote, functional conserved domains in entire genomes by combining relaxed sequence-database searches with fold recognition
- PMID: 21423752
- PMCID: PMC3053371
- DOI: 10.1371/journal.pone.0017568
HMMerThread: detecting remote, functional conserved domains in entire genomes by combining relaxed sequence-database searches with fold recognition
Abstract
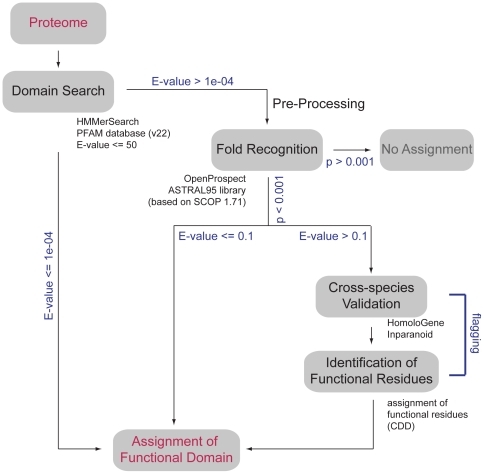
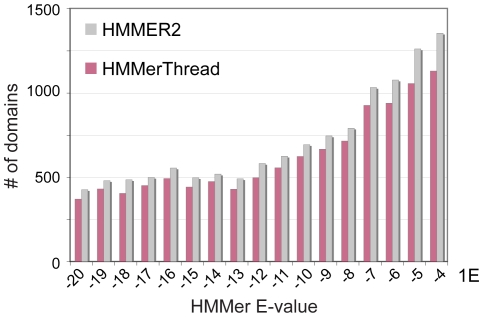
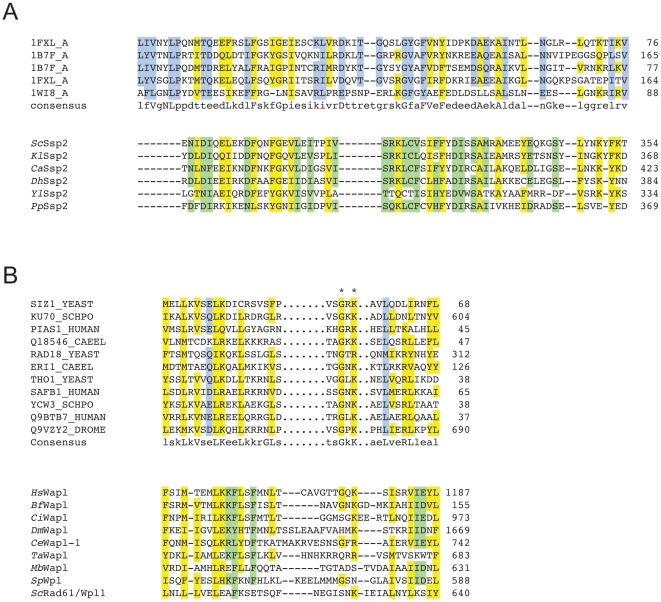
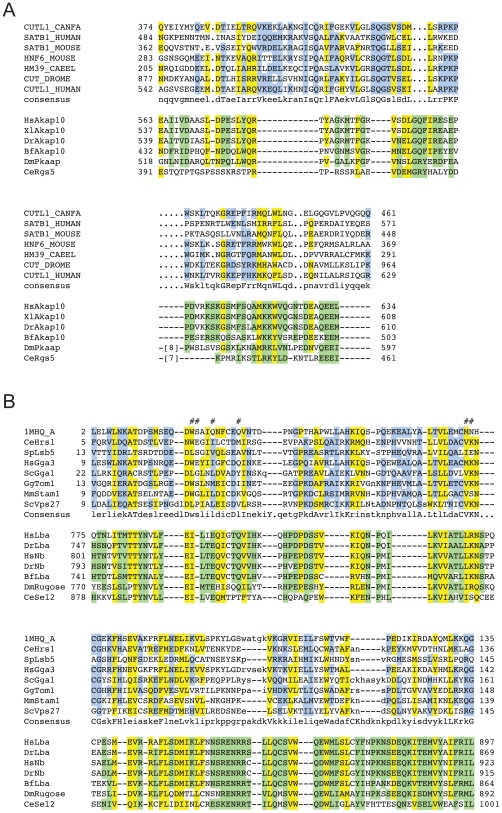
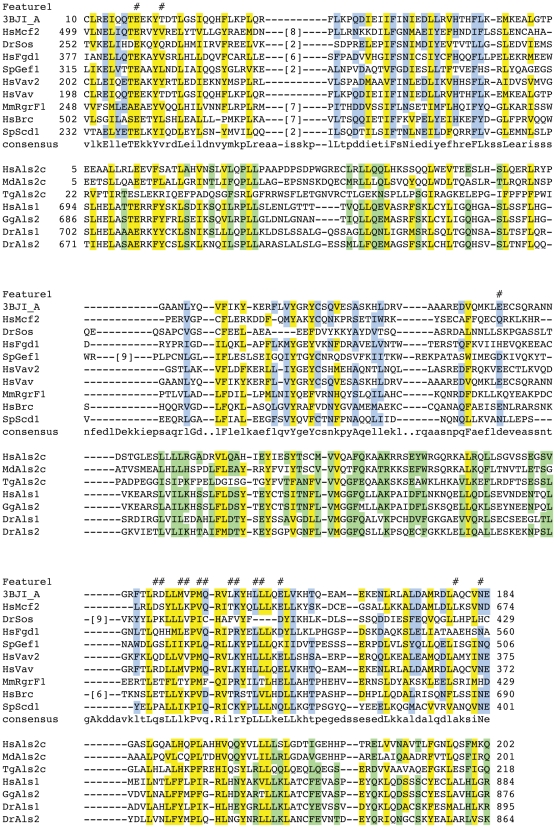
Conserved domains in proteins are one of the major sources of functional information for experimental design and genome-level annotation. Though search tools for conserved domain databases such as Hidden Markov Models (HMMs) are sensitive in detecting conserved domains in proteins when they share sufficient sequence similarity, they tend to miss more divergent family members, as they lack a reliable statistical framework for the detection of low sequence similarity. We have developed a greatly improved HMMerThread algorithm that can detect remotely conserved domains in highly divergent sequences. HMMerThread combines relaxed conserved domain searches with fold recognition to eliminate false positive, sequence-based identifications. With an accuracy of 90%, our software is able to automatically predict highly divergent members of conserved domain families with an associated 3-dimensional structure. We give additional confidence to our predictions by validation across species. We have run HMMerThread searches on eight proteomes including human and present a rich resource of remotely conserved domains, which adds significantly to the functional annotation of entire proteomes. We find ∼4500 cross-species validated, remotely conserved domain predictions in the human proteome alone. As an example, we find a DNA-binding domain in the C-terminal part of the A-kinase anchor protein 10 (AKAP10), a PKA adaptor that has been implicated in cardiac arrhythmias and premature cardiac death, which upon stress likely translocates from mitochondria to the nucleus/nucleolus. Based on our prediction, we propose that with this HLH-domain, AKAP10 is involved in the transcriptional control of stress response. Further remotely conserved domains we discuss are examples from areas such as sporulation, chromosome segregation and signalling during immune response. The HMMerThread algorithm is able to automatically detect the presence of remotely conserved domains in proteins based on weak sequence similarity. Our predictions open up new avenues for biological and medical studies. Genome-wide HMMerThread domains are available at http://vm1-hmmerthread.age.mpg.de.
Conflict of interest statement
Figures

Similar articles
-
ProFAT: a web-based tool for the functional annotation of protein sequences.BMC Bioinformatics. 2006 Oct 23;7:466. doi: 10.1186/1471-2105-7-466. BMC Bioinformatics. 2006. PMID: 17059594 Free PMC article.
-
Improving protein structure similarity searches using domain boundaries based on conserved sequence information.BMC Struct Biol. 2009 May 19;9:33. doi: 10.1186/1472-6807-9-33. BMC Struct Biol. 2009. PMID: 19454035 Free PMC article.
-
Fitting hidden Markov models of protein domains to a target species: application to Plasmodium falciparum.BMC Bioinformatics. 2012 May 1;13:67. doi: 10.1186/1471-2105-13-67. BMC Bioinformatics. 2012. PMID: 22548871 Free PMC article.
-
IgStrand: A universal residue numbering scheme for the immunoglobulin-fold (Ig-fold) to study Ig-proteomes and Ig-interactomes.PLoS Comput Biol. 2025 Apr 14;21(4):e1012813. doi: 10.1371/journal.pcbi.1012813. eCollection 2025 Apr. PLoS Comput Biol. 2025. PMID: 40228037 Free PMC article. Review.
-
Exploiting protein structure data to explore the evolution of protein function and biological complexity.Philos Trans R Soc Lond B Biol Sci. 2006 Mar 29;361(1467):425-40. doi: 10.1098/rstb.2005.1801. Philos Trans R Soc Lond B Biol Sci. 2006. PMID: 16524831 Free PMC article. Review.
Cited by
-
Diversity and prevalence of ANTAR RNAs across actinobacteria.BMC Microbiol. 2021 May 29;21(1):159. doi: 10.1186/s12866-021-02234-x. BMC Microbiol. 2021. PMID: 34051745 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
