

Truncating mutations in NRXN2 and NRXN1 in autism spectrum disorders and schizophrenia
- PMID: 21424692
- PMCID: PMC3204930
- DOI: 10.1007/s00439-011-0975-z
Truncating mutations in NRXN2 and NRXN1 in autism spectrum disorders and schizophrenia
Abstract
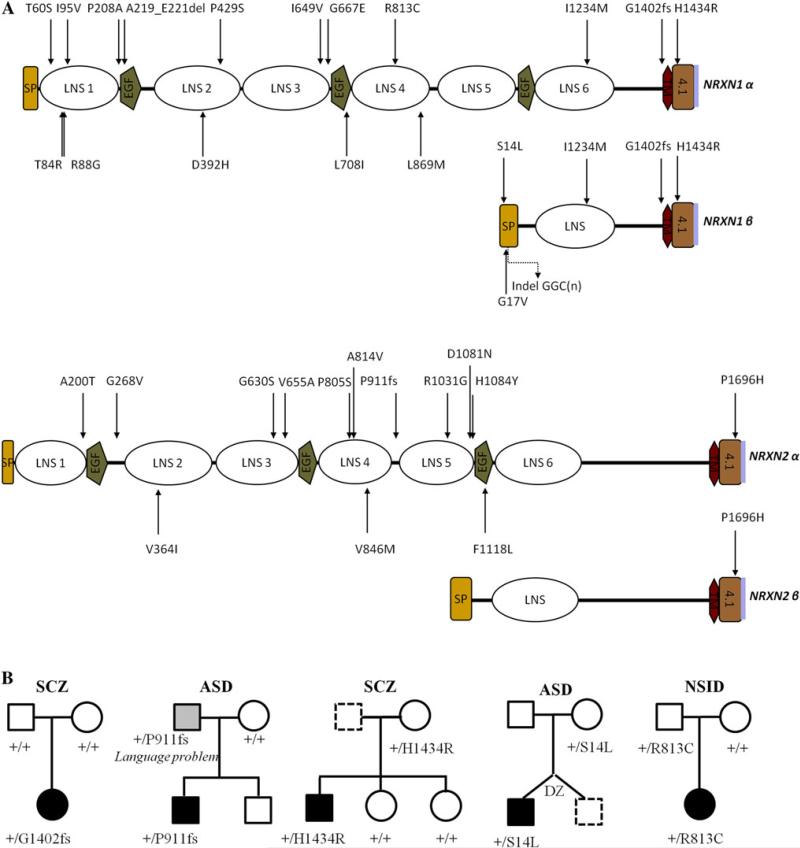
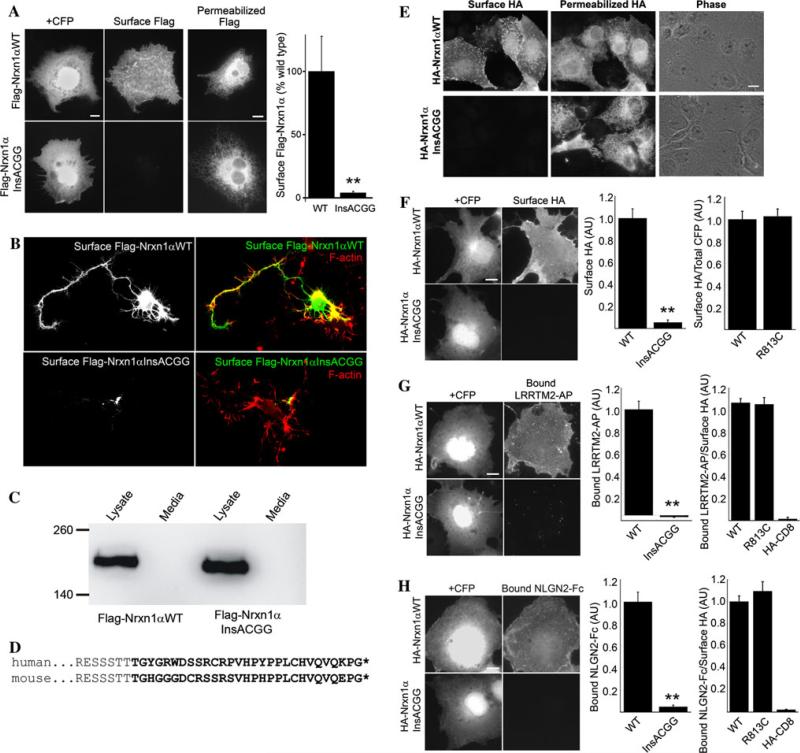
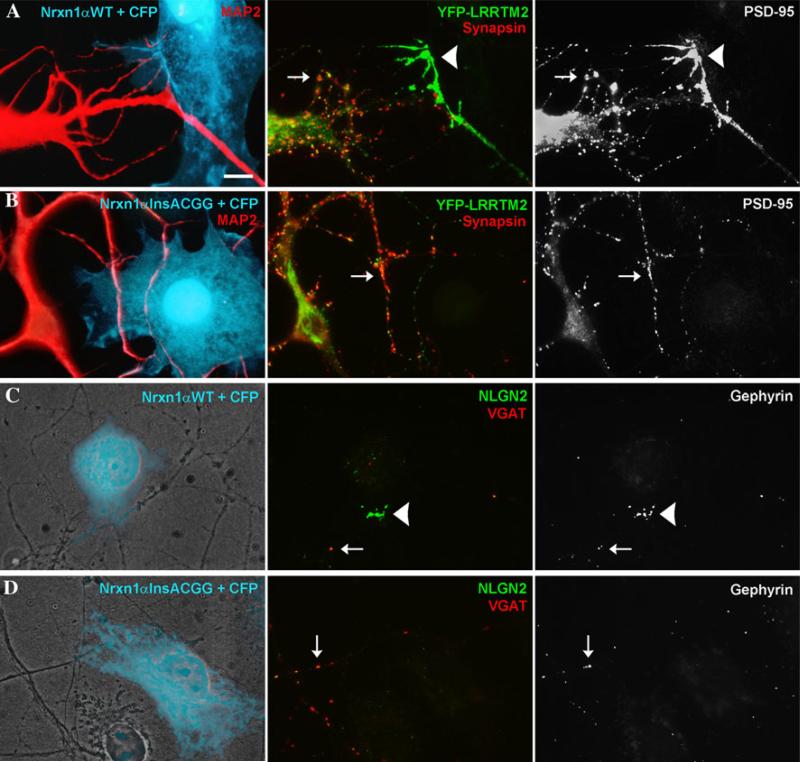
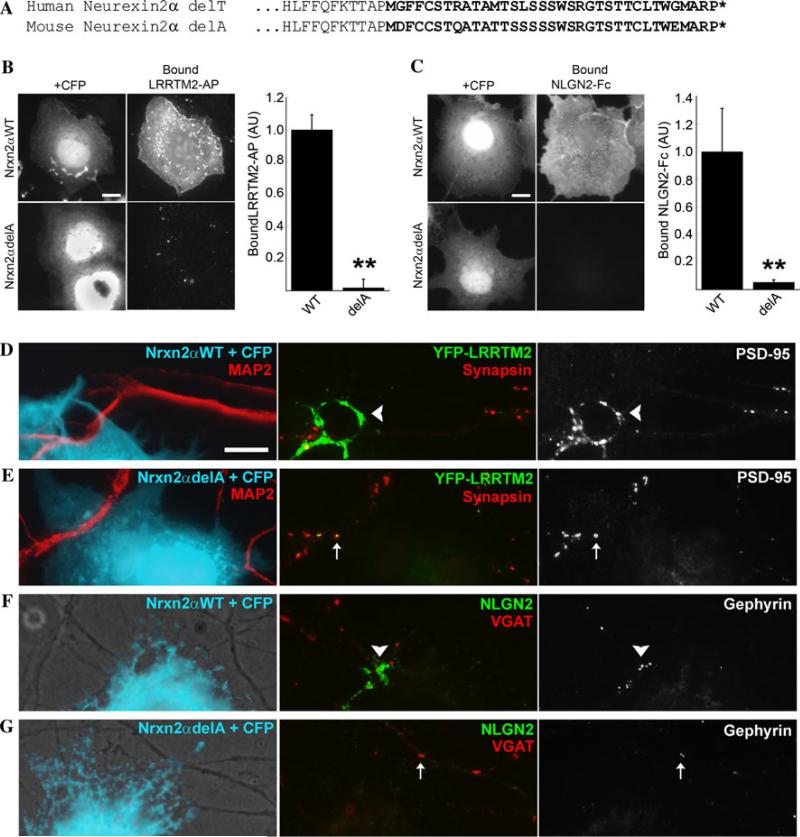
Growing genetic evidence is converging in favor of common pathogenic mechanisms for autism spectrum disorders (ASD), intellectual disability (ID or mental retardation) and schizophrenia (SCZ), three neurodevelopmental disorders affecting cognition and behavior. Copy number variations and deleterious mutations in synaptic organizing proteins including NRXN1 have been associated with these neurodevelopmental disorders, but no such associations have been reported for NRXN2 or NRXN3. From resequencing the three neurexin genes in individuals affected by ASD (n = 142), SCZ (n = 143) or non-syndromic ID (n = 94), we identified a truncating mutation in NRXN2 in a patient with ASD inherited from a father with severe language delay and family history of SCZ. We also identified a de novo truncating mutation in NRXN1 in a patient with SCZ, and other potential pathogenic ASD mutations. These truncating mutations result in proteins that fail to promote synaptic differentiation in neuron coculture and fail to bind either of the established postsynaptic binding partners LRRTM2 or NLGN2 in cell binding assays. Our findings link NRXN2 disruption to the pathogenesis of ASD for the first time and further strengthen the involvement of NRXN1 in SCZ, supporting the notion of a common genetic mechanism in these disorders.
Figures
References
-
- Belmonte MK, Cook EH, Jr, Anderson GM, Rubenstein JL, Greenough WT, Beckel-Mitchener A, Courchesne E, Boulanger LM, Powell SB, Levitt PR, Perry EK, Jiang YH, DeLorey TM, Tierney E. Autism as a disorder of neural information processing: directions for research and targets for therapy. Mol Psychiatry. 2004;9:646–663. - PubMed
-
- Biederer T, Scheiffele P. Mixed-culture assays for analyzing neuronal synapse formation. Nat Protoc. 2007;2:670–676. - PubMed
-
- Biederer T, Sudhof TC. CASK and protein 4.1 support F-actin nucleation on neurexins. J Biol Chem. 2001;276:47869–47876. - PubMed
-
- Boucard AA, Chubykin AA, Comoletti D, Taylor P, Sudhof TC. A splice code for trans-synaptic cell adhesion mediated by binding of neuroligin 1 to alpha- and beta-neurexins. Neuron. 2005;48:229–236. - PubMed
-
- Braff DL, Geyer MA. Sensorimotor gating and schizophrenia. Human and animal model studies. Arch Gen Psychiatry. 1990;47:181–188. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
