

A new phenotype of mitochondrial disease characterized by familial late-onset predominant axial myopathy and encephalopathy
- PMID: 21424749
- PMCID: PMC3098999
- DOI: 10.1007/s00401-011-0818-y
A new phenotype of mitochondrial disease characterized by familial late-onset predominant axial myopathy and encephalopathy
Abstract
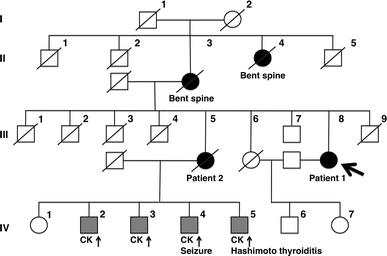
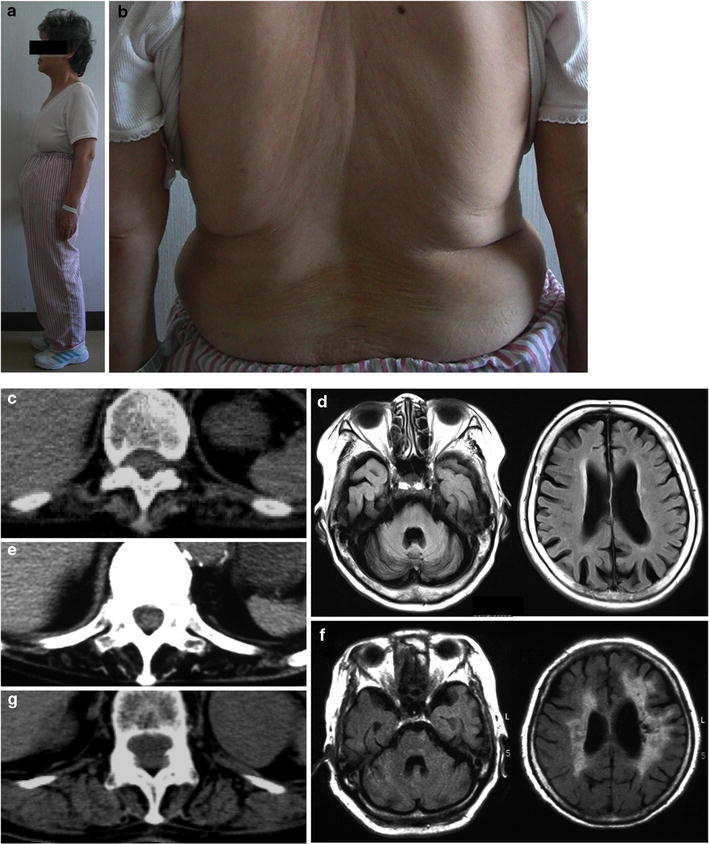
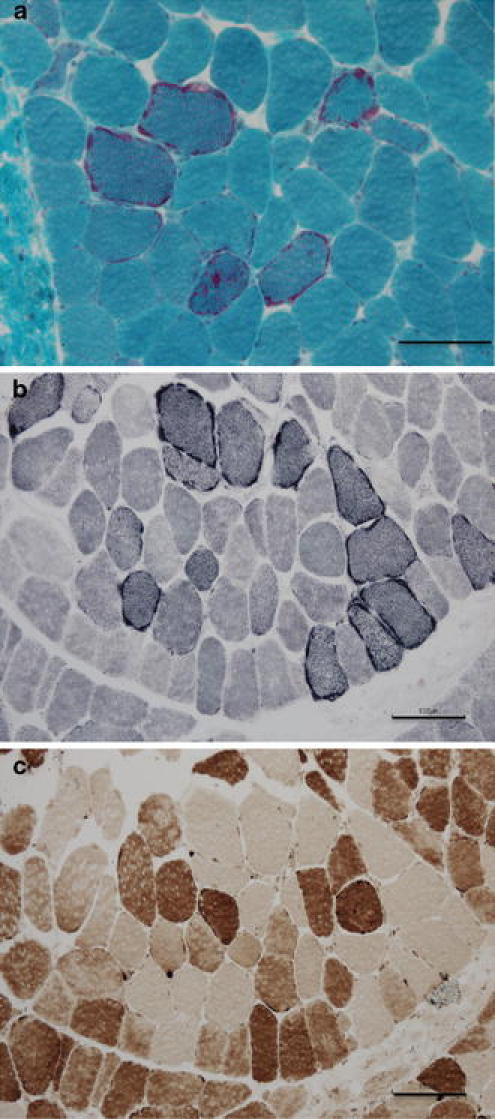
Axial myopathy is a rare neuromuscular disease that is characterized by paraspinal muscle atrophy and abnormal posture, most notably camptocormia (also known as bent spine). The genetic cause of familial axial myopathy is unknown. Described here are the clinical features and cause of late-onset predominant axial myopathy and encephalopathy. A 73-year-old woman presented with a 10-year history of severe paraspinal muscle atrophy and cerebellar ataxia. Her 84-year-old sister also developed late-onset paraspinal muscle atrophy and generalized seizures with encephalopathy. Computed tomography showed severe atrophy and fatty degeneration of their paraspinal muscles. Their mother and maternal aunt also developed bent spines. The existence of many ragged-red fibers and cytochrome c oxidase-negative fibers in the biceps brachii muscle of the proband indicated a mitochondrial abnormality. No significant abnormalities were observed in the respiratory chain enzyme activities; however, the activities of complexes I and IV were relatively low compared with the activities of other complexes. Sequence analysis of the mitochondrial DNA from the muscle revealed a novel heteroplasmic mutation (m.602C>T) in the mitochondrial tRNA(Phe) gene. This familial case of late-onset predominant axial myopathy and encephalopathy may represent a new clinical phenotype of a mitochondrial disease.
Figures


Similar articles
-
Novel mitochondrial transfer RNA(Phe) gene mutation associated with late-onset neuromuscular disease.Arch Neurol. 2006 Jun;63(6):902-5. doi: 10.1001/archneur.63.6.902. Arch Neurol. 2006. PMID: 16769874
-
A novel mutation in the mitochondrial tRNA(Phe) gene associated with mitochondrial myopathy.Neuromuscul Disord. 2004 Jan;14(1):46-50. doi: 10.1016/s0960-8966(03)00168-8. Neuromuscul Disord. 2004. PMID: 14659412
-
Catastrophic presentation of mitochondrial disease due to a mutation in the tRNA(His) gene.Neurology. 2004 Apr 27;62(8):1420-3. doi: 10.1212/01.wnl.0000120667.77372.46. Neurology. 2004. PMID: 15111688
-
Multiple defects of the mitochondrial respiratory chain in a mitochondrial encephalopathy (MERRF): a clinical, biochemical and molecular study.J Neurol Sci. 1991 Mar;102(1):17-24. doi: 10.1016/0022-510x(91)90088-o. J Neurol Sci. 1991. PMID: 1649912 Review.
-
Heteroplasmic mitochondrial DNA mutation in a patient with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes.J Formos Med Assoc. 1995 Jan-Feb;94(1-2):42-7. J Formos Med Assoc. 1995. PMID: 7613232 Review.
Cited by
-
Clinical presentation of axial myopathy in two siblings with HTLV-1 associated myelopathy/tropical spastic paraparesis (HAM/TSP).BMC Neurol. 2015 Feb 28;15:18. doi: 10.1186/s12883-015-0275-7. BMC Neurol. 2015. PMID: 25884435 Free PMC article.
-
Pathophysiological Concepts and Treatment of Camptocormia.J Parkinsons Dis. 2016 Jun 16;6(3):485-501. doi: 10.3233/JPD-160836. J Parkinsons Dis. 2016. PMID: 27314757 Free PMC article. Review.
-
A proposed consensus panel of organisms for determining evolutionary conservation of mt-tRNA point mutations.Mitochondrion. 2012 Sep;12(5):533-8. doi: 10.1016/j.mito.2012.06.009. Epub 2012 Jul 7. Mitochondrion. 2012. PMID: 22781547 Free PMC article.
-
Camptocormia and shuffling gait due to a novel MT-TV mutation: Diagnostic pitfalls.Neurol Genet. 2017 Apr 5;3(3):e147. doi: 10.1212/NXG.0000000000000147. eCollection 2017 Jun. Neurol Genet. 2017. PMID: 28396884 Free PMC article. No abstract available.
-
Axial mitochondrial myopathy in a patient with rapidly progressive adult-onset scoliosis.Acta Neuropathol Commun. 2014 Sep 16;2:137. doi: 10.1186/s40478-014-0137-3. Acta Neuropathol Commun. 2014. PMID: 25223649 Free PMC article.
References
-
- Berio A, Piazzi A. A case of Kearns-Sayre syndrome with autoimmune thyroiditis and possible Hashimoto encephalopathy. Panminerva Med. 2002;44:265–269. - PubMed
-
- Bernier FP, Boneh A, Dennett X, Chow CW, Cleary MA, Thorburn DR. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology. 2002;59:1406–1411. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
