

Hexokinase regulates Bax-mediated mitochondrial membrane injury following ischemic stress
- PMID: 21430642
- PMCID: PMC3361076
- DOI: 10.1038/ki.2010.532
Hexokinase regulates Bax-mediated mitochondrial membrane injury following ischemic stress
Abstract
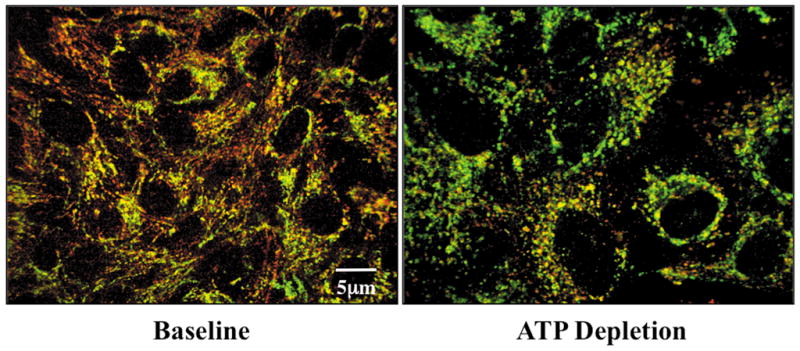
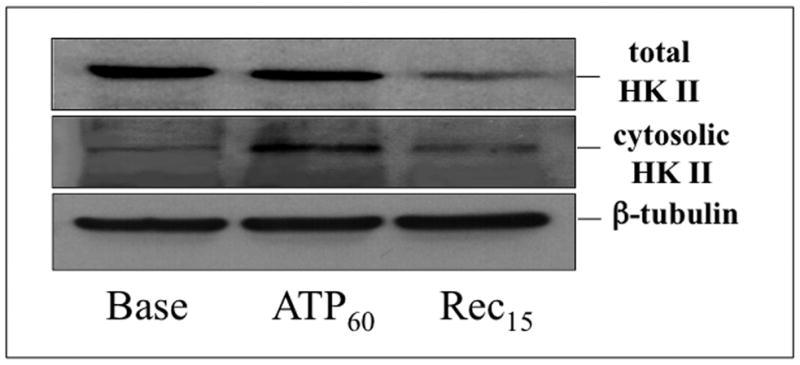
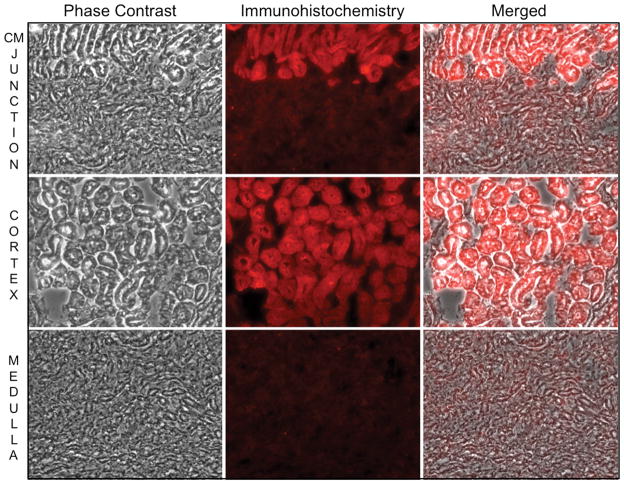
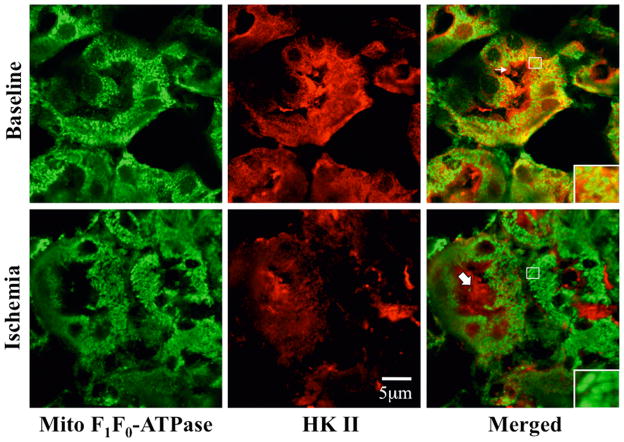
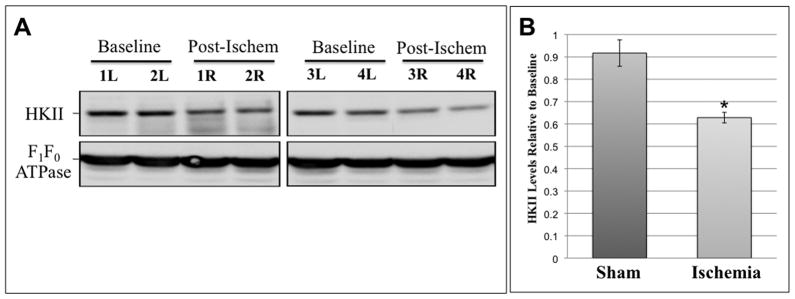
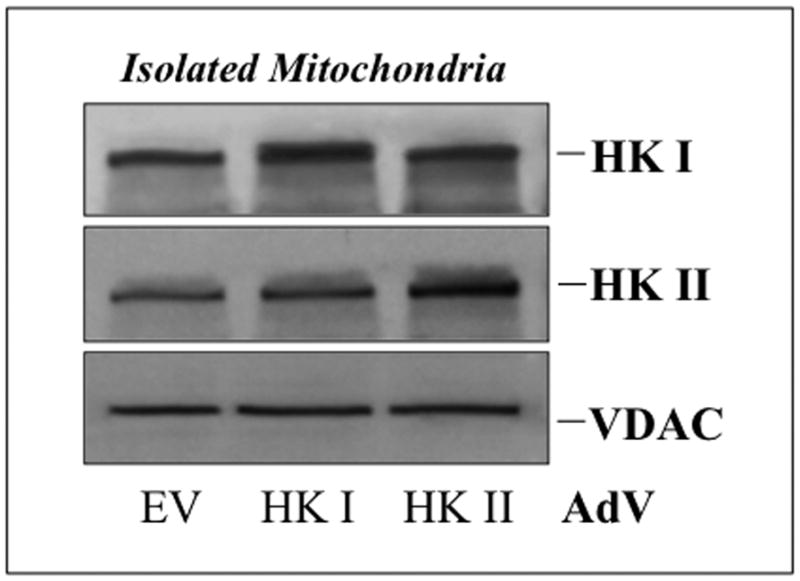
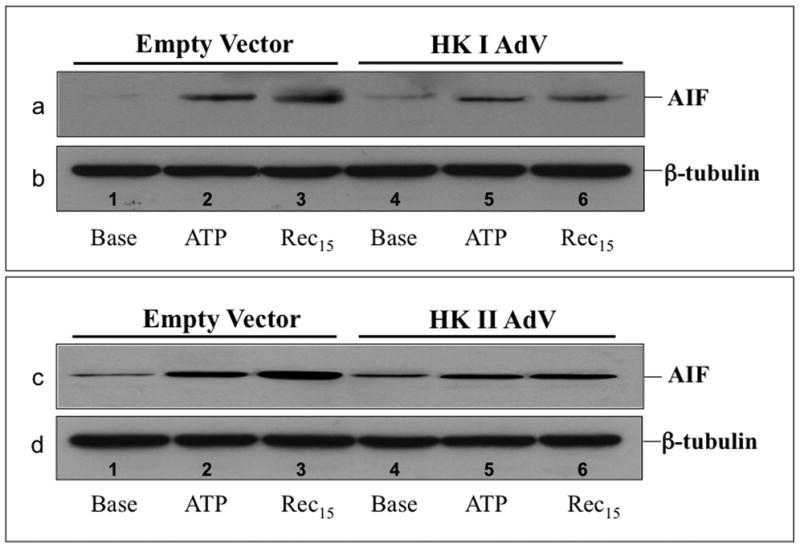
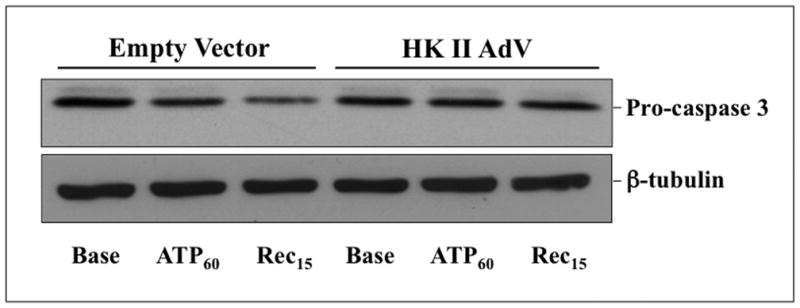
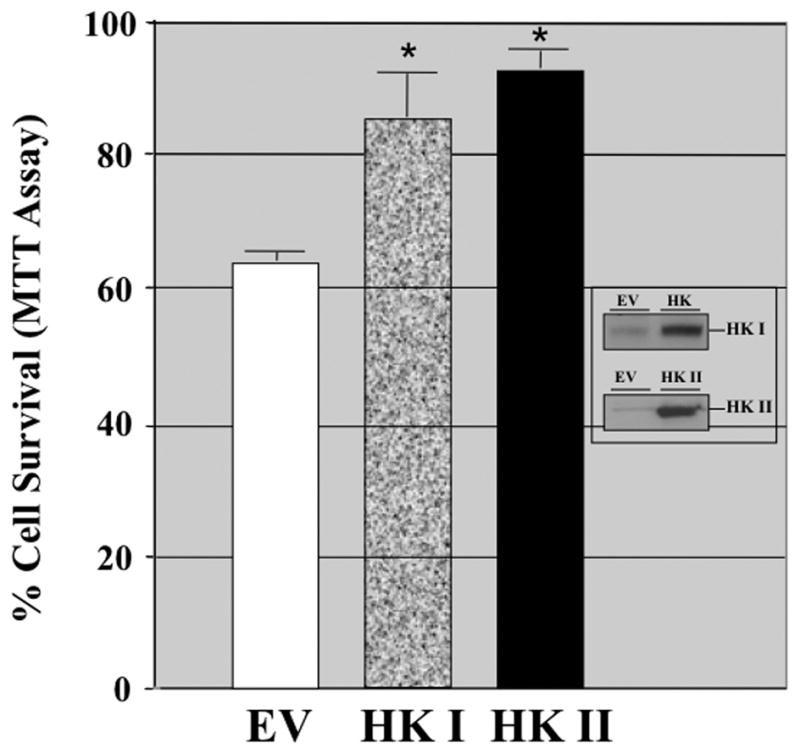
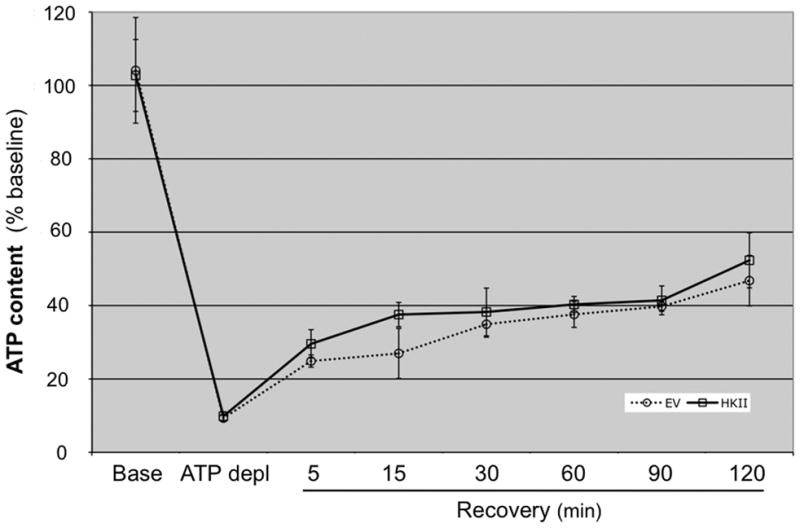
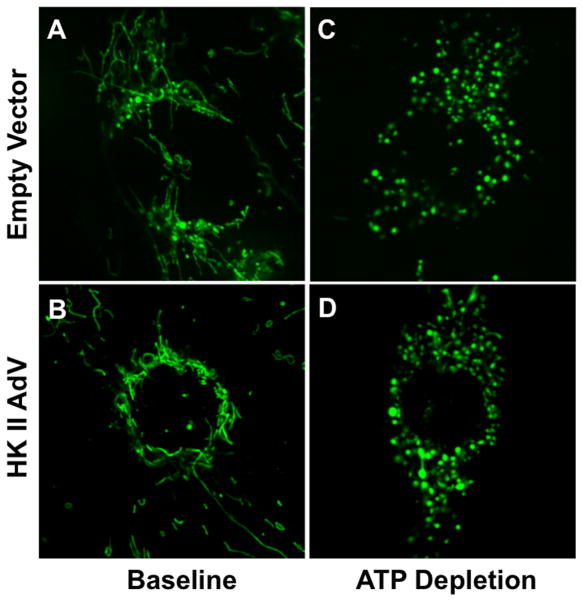
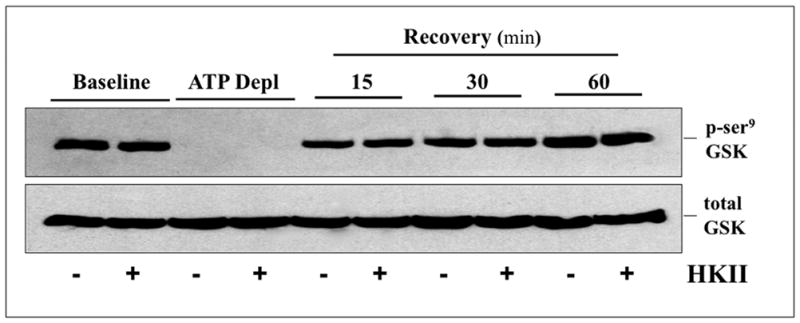
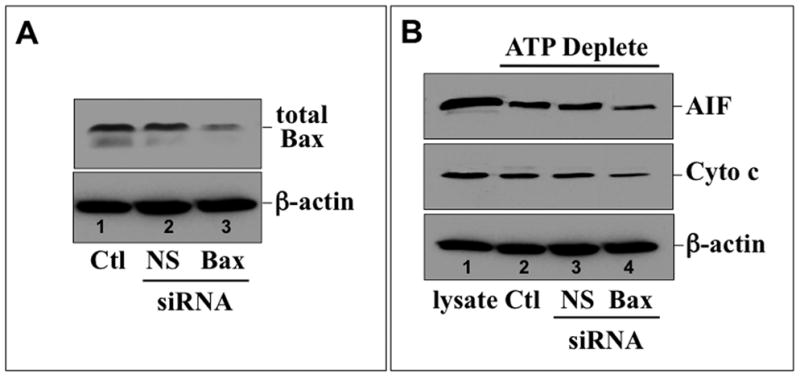
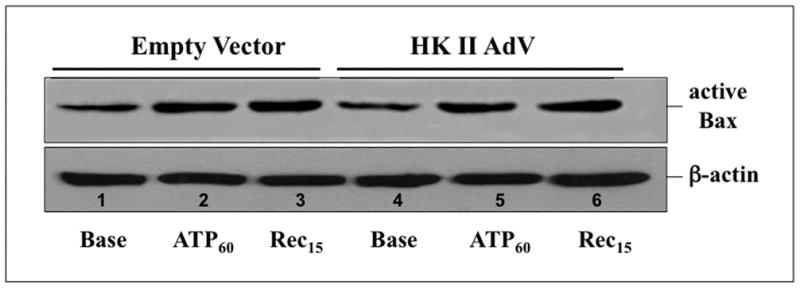
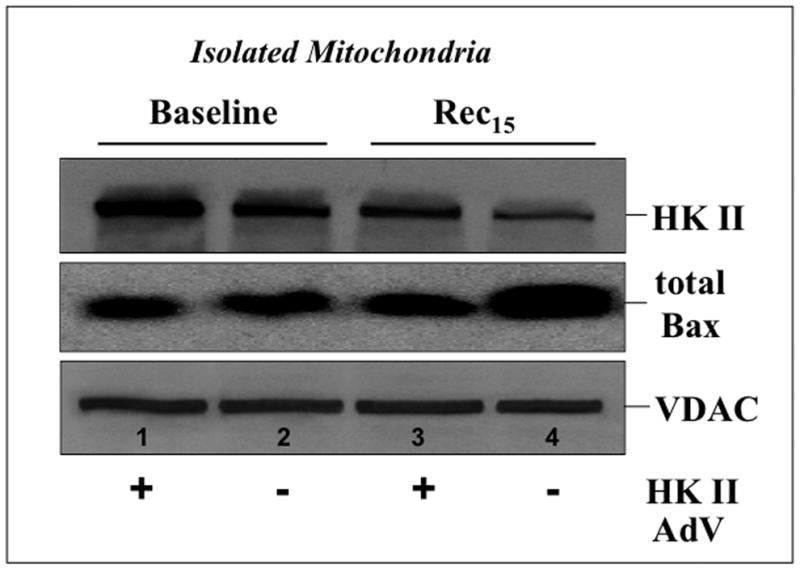
Hexokinase (HK), the rate-limiting enzyme in glycolysis, controls cell survival by promoting metabolism and/or inhibiting apoptosis. Since HK isoforms I and II have mitochondrial targeting sequences, we attempted to separate the protective effects of HK on cell metabolism from those on apoptosis. We exposed renal epithelial cells to metabolic stress causing ATP depletion in the absence of glucose and found that this activated glycogen synthase kinase 3β (GSK3β) and Bax caused mitochondrial membrane injury and apoptosis. ATP depletion led to a progressive HK II dissociation from mitochondria, released mitochondrial apoptosis inducing factor and cytochrome c into the cytosol, activated caspase-3, and reduced cell survival. Compared with control, adenoviral-mediated HK I or II overexpression improved cell survival following stress, but did not prevent GSK3β or Bax activation, improve ATP content, or reduce mitochondrial fragmentation. HK I or HK II overexpression increased mitochondria-associated isoform-specific HK content, and decreased mitochondrial membrane injury and apoptosis after stress. In vivo, HK II localized exclusively to the proximal tubule. Ischemia reduced total renal HK II content and dissociated HK II from proximal tubule mitochondria. In cells overexpressing HK II, Bax and HK II did not interact before or after stress. While the mechanism by which HK antagonizes Bax-mediated apoptosis is unresolved by these studies, one possible scenario is that the two proteins compete for a common binding site on the outer mitochondrial membrane.
Conflict of interest statement
The authors have no competing financial interests.
Figures
Comment in
-
Hexokinase: a novel sugar kinase coupled to renal epithelial cell survival.Kidney Int. 2011 Jun;79(11):1163-5. doi: 10.1038/ki.2011.20. Kidney Int. 2011. PMID: 21566638
References
-
- Bryson JM, Coy PE, Gottlob K, Hay N, et al. Increased hexokinase activity, of either ectopic or endogenous origin, protects renal epithelial cells against acute oxidant-induced cell death. J Biol Chem. 2002;277:11392–11400. - PubMed
-
- Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol. 2003;206:2049–2057. - PubMed
-
- Ballatori N, Cohen JJ. Intracellular distribution of hexokinase in the tissue zones of rat kidney. Biochim Biophys Acta. 1981;657:448–456. - PubMed
-
- Vandewalle A, Wirthensohn G, Heidrich HG, Guder WG. Distribution of hexokinase and phosphoenolpyruvate carboxykinase along the rabbit nephron. Am J Physiol. 1981;240:F492–500. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
