

Diagnosis and management of glutaric aciduria type I--revised recommendations
- PMID: 21431622
- PMCID: PMC3109243
- DOI: 10.1007/s10545-011-9289-5
Diagnosis and management of glutaric aciduria type I--revised recommendations
Abstract
Glutaric aciduria type I (synonym, glutaric acidemia type I) is a rare organic aciduria. Untreated patients characteristically develop dystonia during infancy resulting in a high morbidity and mortality. The neuropathological correlate is striatal injury which results from encephalopathic crises precipitated by infectious diseases, immunizations and surgery during a finite period of brain development, or develops insidiously without clinically apparent crises. Glutaric aciduria type I is caused by inherited deficiency of glutaryl-CoA dehydrogenase which is involved in the catabolic pathways of L-lysine, L-hydroxylysine and L-tryptophan. This defect gives rise to elevated glutaric acid, 3-hydroxyglutaric acid, glutaconic acid, and glutarylcarnitine which can be detected by gas chromatography/mass spectrometry (organic acids) or tandem mass spectrometry (acylcarnitines). Glutaric aciduria type I is included in the panel of diseases that are identified by expanded newborn screening in some countries. It has been shown that in the majority of neonatally diagnosed patients striatal injury can be prevented by combined metabolic treatment. Metabolic treatment that includes a low lysine diet, carnitine supplementation and intensified emergency treatment during acute episodes of intercurrent illness should be introduced and monitored by an experienced interdisciplinary team. However, initiation of treatment after the onset of symptoms is generally not effective in preventing permanent damage. Secondary dystonia is often difficult to treat, and the efficacy of available drugs cannot be predicted precisely in individual patients. The major aim of this revision is to re-evaluate the previous diagnostic and therapeutic recommendations for patients with this disease and incorporate new research findings into the guideline.
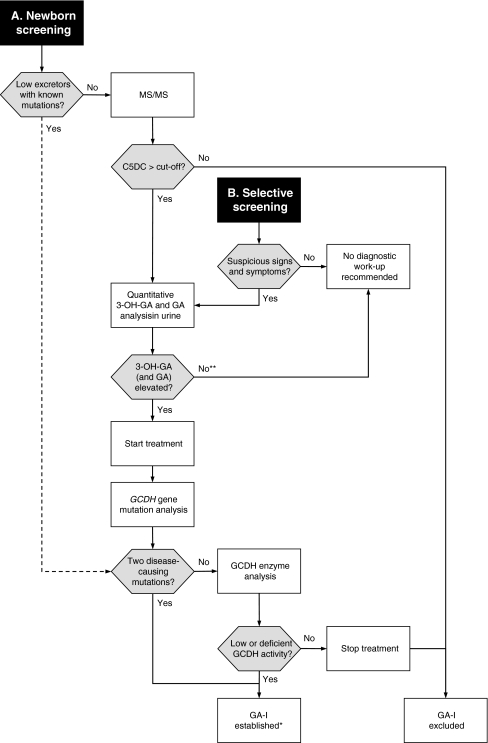
Figures
References
-
- Al-Dirbashi OY, Jacob M, Al-Amoudi M, Al-Odaib A, Al-Kahtani, El-Badaoui F, Rashed MS. Quantification of glutaric and 3-hydroxyglutaric acids in urine of glutaric acidemia type I patients by HPLC with intramolecular excimer-forming fluorescence derivatization. Clin Chim Acta. 2005;359:179–188. doi: 10.1016/j.cccn.2005.03.048. - DOI - PubMed
-
- Bähr O, Mader I, Zschocke J, Dichgans J, Schulz JB. Adult onset glutaric aciduria type I presenting with leukoencephalopathy. Neurology. 2002;59:1802–1804. - PubMed
-
- Baric I, Wagner L, Feyh P, Liesert M, Buckel W, Hoffmann GF. Sensitivity of free and total glutaric and 3-hydroxyglutaric acid measurement by stable isotope dilution assays for the diagnosis of glutaric aciduria type I. J Inherit Metab Dis. 1999;22:867–882. doi: 10.1023/A:1005683222187. - DOI - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
