

Meta-analysis of gene expression microarrays with missing replicates
- PMID: 21435268
- PMCID: PMC3224118
- DOI: 10.1186/1471-2105-12-84
Meta-analysis of gene expression microarrays with missing replicates
Abstract
Background: Many different microarray experiments are publicly available today. It is natural to ask whether different experiments for the same phenotypic conditions can be combined using meta-analysis, in order to increase the overall sample size. However, some genes are not measured in all experiments, hence they cannot be included or their statistical significance cannot be appropriately estimated in traditional meta-analysis. Nonetheless, these genes, which we refer to as incomplete genes, may also be informative and useful.
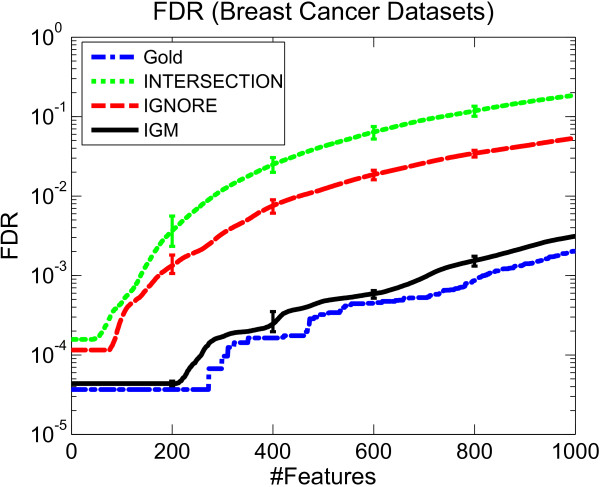
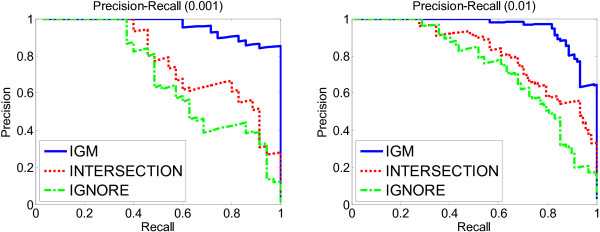
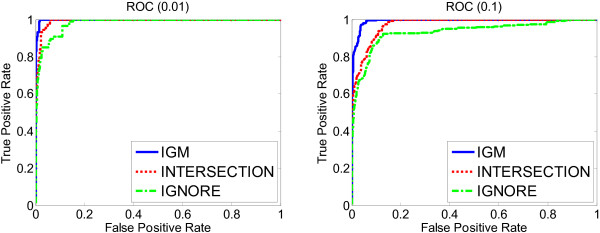
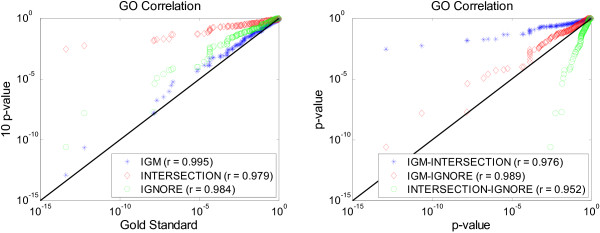
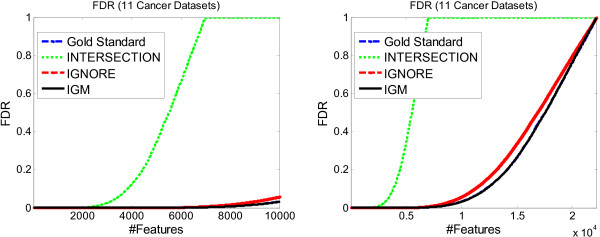
Results: We propose a meta-analysis framework, called "Incomplete Gene Meta-analysis", which can include incomplete genes by imputing the significance of missing replicates, and computing a meta-score for every gene across all datasets. We demonstrate that the incomplete genes are worthy of being included and our method is able to appropriately estimate their significance in two groups of experiments. We first apply the Incomplete Gene Meta-analysis and several comparable methods to five breast cancer datasets with an identical set of probes. We simulate incomplete genes by randomly removing a subset of probes from each dataset and demonstrate that our method consistently outperforms two other methods in terms of their false discovery rate. We also apply the methods to three gastric cancer datasets for the purpose of discriminating diffuse and intestinal subtypes.
Conclusions: Meta-analysis is an effective approach that identifies more robust sets of differentially expressed genes from multiple studies. The incomplete genes that mainly arise from the use of different platforms may also have statistical and biological importance but are ignored or are not appropriately involved by previous studies. Our Incomplete Gene Meta-analysis is able to incorporate the incomplete genes by estimating their significance. The results on both breast and gastric cancer datasets suggest that the highly ranked genes and associated GO terms produced by our method are more significant and biologically meaningful according to the previous literature.
Figures
Similar articles
-
Robust microarray meta-analysis identifies differentially expressed genes for clinical prediction.ScientificWorldJournal. 2012;2012:989637. doi: 10.1100/2012/989637. Epub 2012 Dec 18. ScientificWorldJournal. 2012. PMID: 23365541 Free PMC article.
-
Statistical Test of Expression Pattern (STEPath): a new strategy to integrate gene expression data with genomic information in individual and meta-analysis studies.BMC Bioinformatics. 2011 Apr 11;12:92. doi: 10.1186/1471-2105-12-92. BMC Bioinformatics. 2011. PMID: 21481242 Free PMC article.
-
MAID : an effect size based model for microarray data integration across laboratories and platforms.BMC Bioinformatics. 2008 Jul 10;9:305. doi: 10.1186/1471-2105-9-305. BMC Bioinformatics. 2008. PMID: 18616827 Free PMC article.
-
Consistent Differential Expression Pattern (CDEP) on microarray to identify genes related to metastatic behavior.BMC Bioinformatics. 2011 Nov 11;12:438. doi: 10.1186/1471-2105-12-438. BMC Bioinformatics. 2011. PMID: 22078224 Free PMC article.
-
A meta-data based method for DNA microarray imputation.BMC Bioinformatics. 2007 Mar 29;8:109. doi: 10.1186/1471-2105-8-109. BMC Bioinformatics. 2007. PMID: 17394658 Free PMC article.
Cited by
-
MAAMD: a workflow to standardize meta-analyses and comparison of affymetrix microarray data.BMC Bioinformatics. 2014 Mar 12;15:69. doi: 10.1186/1471-2105-15-69. BMC Bioinformatics. 2014. PMID: 24621103 Free PMC article.
-
Integrated genomic approaches identify major pathways and upstream regulators in late onset Alzheimer's disease.Sci Rep. 2015 Jul 23;5:12393. doi: 10.1038/srep12393. Sci Rep. 2015. PMID: 26202100 Free PMC article.
References
-
- Hedges LV, Olkin I. Statistical Methods for Meta-Analysis. Academic Press. San Diego, CA, USA; 1985.
-
- Rhodes DR, Barrette TR, Rubin MA, Ghosh D, Chinnaiyan AM. Meta-Analysis of Microarrays: Interstudy Validation of Gene Expression Profiles Reveals Pathway Dysregulation in Prostate Cancer. Cancer Research. 2002;62(15):4427–4433. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
