

Phagocytic clearance in neurodegeneration
- PMID: 21435432
- PMCID: PMC3078427
- DOI: 10.1016/j.ajpath.2010.12.051
Phagocytic clearance in neurodegeneration
Abstract
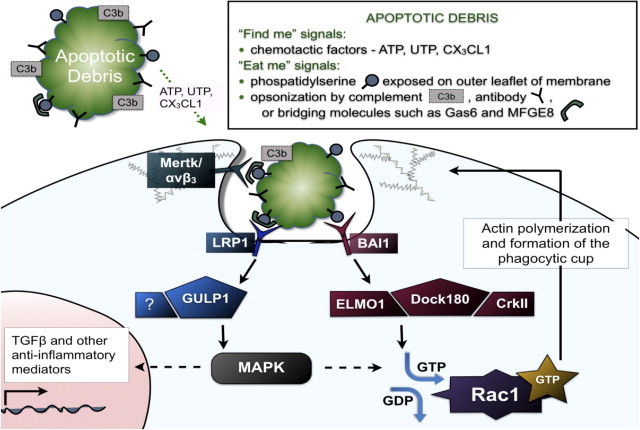
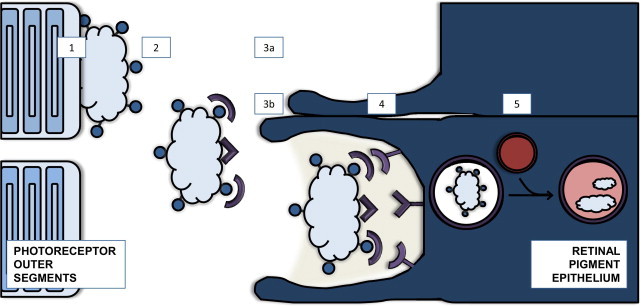
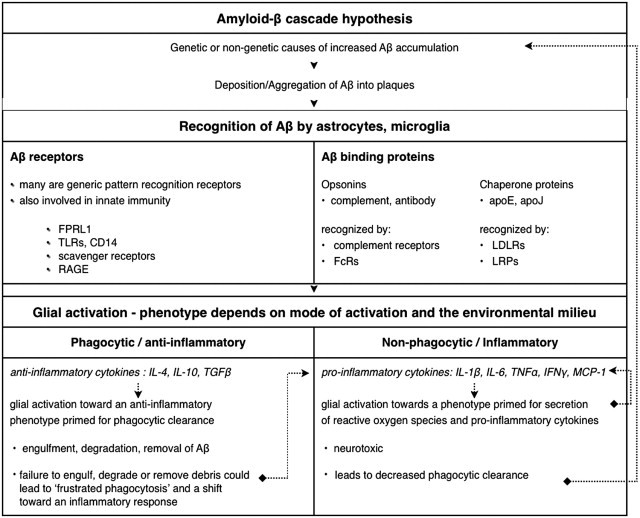
The cellular and molecular mechanisms of phagocytic clearance of apoptotic cells and debris have been intensely studied in invertebrate model organisms and in the mammalian immune system. This evolutionarily conserved process serves multiple purposes. Uncleared debris from dying cells or aggregated proteins can be toxic and may trigger exaggerated inflammatory responses. Even though apoptotic cell death and debris accumulation are key features of neurodegenerative diseases, relatively little attention has been paid to this important homeostatic function in the central nervous system (CNS). This review attempts to summarize our knowledge of phagocytic clearance in the CNS, with a focus on retinal degeneration, forms of which are caused by mutations in genes within known phagocytic pathways, and on Alzheimer's disease (AD). Interest in phagocytic clearance mechanisms in AD was stimulated by the discovery that immunization could promote phagocytic clearance of amyloid-β; however, much less is known about clearance of neuronal and synaptic corpses in AD and other neurodegenerative diseases. Because the regulation of phagocytic activity is intertwined with cytokine signaling, this review also addresses the relationships among CNS inflammation, glial responses, and phagocytic clearance.
Copyright © 2011 American Society for Investigative Pathology. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Cahoy J.D., Emery B., Kaushal A., Foo L.C., Zamanian J.L., Christopherson K.S., Xing Y., Lubischer J.L., Krieg P.A., Krupenko S.A., Thompson W.J., Barres B.A. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. - PMC - PubMed
-
- DeWitt D.A., Perry G., Cohen M., Doller C., Silver J. Astrocytes regulate microglial phagocytosis of senile plaque cores of Alzheimer's disease. Exp Neurol. 1998;149:329–340. - PubMed
-
- Takuma K., Fang F., Zhang W., Yan S., Fukuzaki E., Du H., Sosunov A., McKhann G., Funatsu Y., Nakamichi N., Nagai T., Mizoguchi H., Ibi D., Hori O., Ogawa S., Stern D.M., Yamada K., Yan S.S. RAGE-mediated signaling contributes to intraneuronal transport of amyloid-beta and neuronal dysfunction. Proc Natl Acad Sci USA. 2009;106:20021–20026. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
