

Therapeutic approaches to muscular dystrophy
- PMID: 21436158
- PMCID: PMC3095062
- DOI: 10.1093/hmg/ddr105
Therapeutic approaches to muscular dystrophy
Abstract
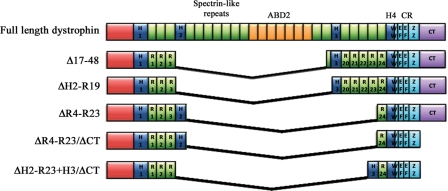
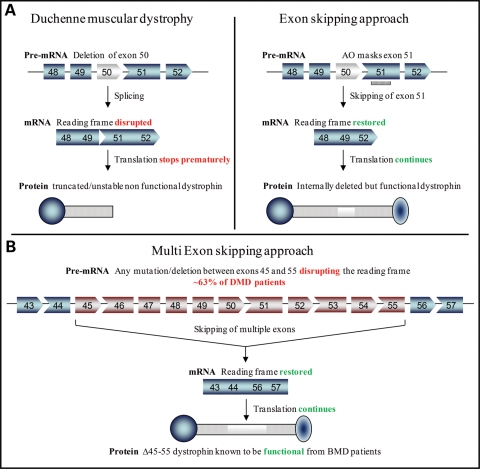
Muscular dystrophies are a heterogeneous group of genetic disorders characterized by muscle weakness and wasting. Duchenne muscular dystrophy (DMD) is the most common and severe form of muscular dystrophy, and although the molecular mechanisms of the disease have been extensively investigated since the discovery of the gene in 1986, there is currently no effective treatment. However, new gene-based therapies have recently emerged with particular noted advances in using conventional gene replacement strategies, RNA-based technology and pharmacological approaches. While the proof of principle has been demonstrated in animal models, several clinical trials have recently been undertaken to investigate the feasibility of these strategies in patients. In particular, antisense-mediated exon skipping has shown encouraging results and holds promise for the treatment of dystrophic muscle. Here, we summarize the recent progress in therapeutic approaches to muscular dystrophies, with an emphasis on gene therapy and exon skipping for DMD.
Figures
References
-
- Campbell K.P. Three muscular dystrophies: loss of cytoskeleton-extracellular matrix linkage. Cell. 1995;80:675–679. doi:10.1016/0092-8674(95)90344-5. - DOI - PubMed
-
- Kafri T., Blomer U., Peterson D.A., Gage F.H., Verma I.M. Sustained expression of genes delivered directly into liver and muscle by lentiviral vectors. Nat. Genet. 1997;17:314–317. doi:10.1038/ng1197-314. - DOI - PubMed
-
- Li S., Kimura E., Fall B.M., Reyes M., Angello J.C., Welikson R., Hauschka S.D., Chamberlain J.S. Stable transduction of myogenic cells with lentiviral vectors expressing a minidystrophin. Gene Ther. 2005;12:1099–1108. doi:10.1038/sj.gt.3302505. - DOI - PubMed
-
- Beard B.C., Keyser K.A., Trobridge G.D., Peterson L.J., Miller D.G., Jacobs M., Kaul R., Kiem H.P. Unique integration profiles in a canine model of long-term repopulating cells transduced with gammaretrovirus, lentivirus, or foamy virus. Hum. Gene Ther. 2007;18:423–434. doi:10.1089/hum.2007.011. - DOI - PubMed
-
- Hacein-Bey-Abina S., Von Kalle C., Schmidt M., McCormack M.P., Wulffraat N., Leboulch P., Lim A., Osborne C.S., Pawliuk R., Morillon E., et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi:10.1126/science.1088547. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
