

Gene-environment interactions: key to unraveling the mystery of Parkinson's disease
- PMID: 21439347
- PMCID: PMC3098527
- DOI: 10.1016/j.pneurobio.2011.03.005
Gene-environment interactions: key to unraveling the mystery of Parkinson's disease
Abstract
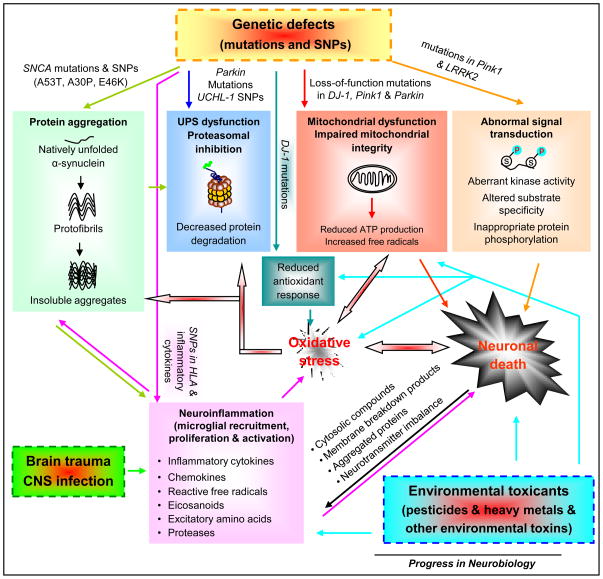
Parkinson's disease (PD) is the second most common neurodegenerative disease. The gradual, irreversible loss of dopamine neurons in the substantia nigra is the signature lesion of PD. Clinical symptoms of PD become apparent when 50-60% of nigral dopamine neurons are lost. PD progresses insidiously for 5-7 years (preclinical period) and then continues to worsen even under the symptomatic treatment. To determine what triggers the disease onset and what drives the chronic, self-propelling neurodegenerative process becomes critical and urgent, since lack of such knowledge impedes the discovery of effective treatments to retard PD progression. At present, available therapeutics only temporarily relieve PD symptoms. While the identification of causative gene defects in familial PD uncovers important genetic influences in this disease, the majority of PD cases are sporadic and idiopathic. The current consensus suggests that PD develops from multiple risk factors including aging, genetic predisposition, and environmental exposure. Here, we briefly review research on the genetic and environmental causes of PD. We also summarize very recent genome-wide association studies on risk gene polymorphisms in the emergence of PD. We highlight the new converging evidence on gene-environment interplay in the development of PD with an emphasis on newly developed multiple-hit PD models involving both genetic lesions and environmental triggers.
Published by Elsevier Ltd.
Figures
References
-
- Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. - PubMed
-
- Ahn TB, Kim SY, Kim JY, Park SS, Lee DS, Min HJ, Kim YK, Kim SE, Kim JM, Kim HJ, Cho J, Jeon BS. alpha-Synuclein gene duplication is present in sporadic Parkinson disease. Neurology. 2008;70:43–49. - PubMed
-
- Alam M, Schmidt WJ. Rotenone destroys dopaminergic neurons and induces parkinsonian symptoms in rats. Behav Brain Res. 2002;136:317–324. - PubMed
-
- Andres-Mateos E, Mejias R, Sasaki M, Li X, Lin BM, Biskup S, Zhang L, Banerjee R, Thomas B, Yang L, Liu G, Beal MF, Huso DL, Dawson TM, Dawson VL. Unexpected lack of hypersensitivity in LRRK2 knock-out mice to MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) J Neurosci. 2009;29:15846–15850. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
