

A solid phase extraction-based platform for rapid phosphoproteomic analysis
- PMID: 21440633
- PMCID: PMC3156280
- DOI: 10.1016/j.ymeth.2011.03.008
A solid phase extraction-based platform for rapid phosphoproteomic analysis
Abstract
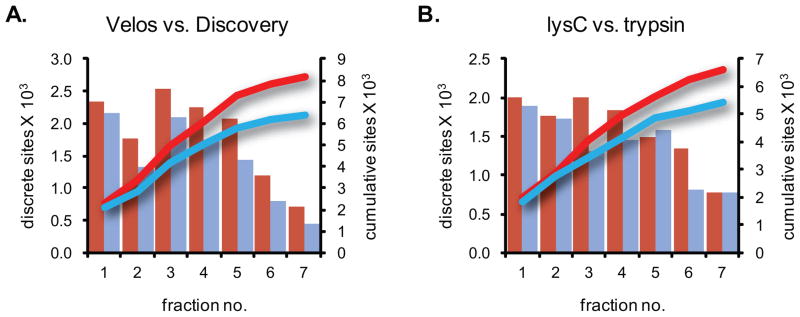
Protein phosphorylation is among the most common and intensely studied post-translational protein modification. It plays crucial roles in virtually all cellular processes and has been implicated in numerous human diseases, including cancer. Traditional biochemical and genetic methods for identifying and monitoring sites of phosphorylation are laborious and slow and in recent years have largely been replaced by mass spectrometric analysis. Improved methods for phosphopeptide enrichment coupled with faster and more sensitive mass spectrometers have led to an explosion in the size of phosphoproteomic datasets. However, wider application of these methods is limited by equipment costs and the resultant high demand for instrument time as well as by a technology gap between biologists and mass spectrometrists. Here we describe a modified two-step enrichment strategy that employs lysC digestion and step elution from self-packed strong cation exchange (SCX) solid phase extraction (SPE) columns followed by immobilized metal ion affinity chromatography (IMAC) and LC-MS/MS analysis using a hybrid LTQ Orbitrap Velos mass spectrometer. The SCX procedure does not require an HPLC system, demands little expertise, and because multiple samples can be processed in parallel, can provide a large savings of time and labor. We demonstrate this method in conjunction with stable isotope labeling to quantitate peptides harboring >8000 unique phosphorylation sites in yeast in 12h of instrument analysis time and examine the impact of enzyme choice and instrument platform.
Copyright © 2011. Published by Elsevier Inc.
Figures



References
-
- Hunter T. Signaling--2000 and beyond. Cell. 2000;100(1):113–127. - PubMed
-
- Yates JR, 3rd, Eng JK, McCormack AL, Schieltz D. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal Chem. 1995;67(8):1426–1436. - PubMed
-
- Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20(18):3551–3567. - PubMed
-
- Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4(3):207–214. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
