

Review
doi: 10.1101/cshperspect.a007476.
Huntington's Disease
Affiliations
- PMID: 21441583
- PMCID: PMC3098678
- DOI: 10.1101/cshperspect.a007476
Item in Clipboard
Review
Huntington's Disease
Cold Spring Harb Perspect Biol.
.
Abstract
Huntington's disease (HD) is the most common inherited neurodegenerative disease and is characterized by uncontrolled excessive motor movements and cognitive and emotional deficits. The mutation responsible for HD leads to an abnormally long polyglutamine (polyQ) expansion in the huntingtin (Htt) protein, which confers one or more toxic functions to mutant Htt leading to neurodegeneration. The polyQ expansion makes Htt prone to aggregate and accumulate, and manipulations that mitigate protein misfolding or facilitate the clearance of misfolded proteins tend to slow disease progression in HD models. This article will focus on HD and the evidence that it is a conformational disease.
Figures
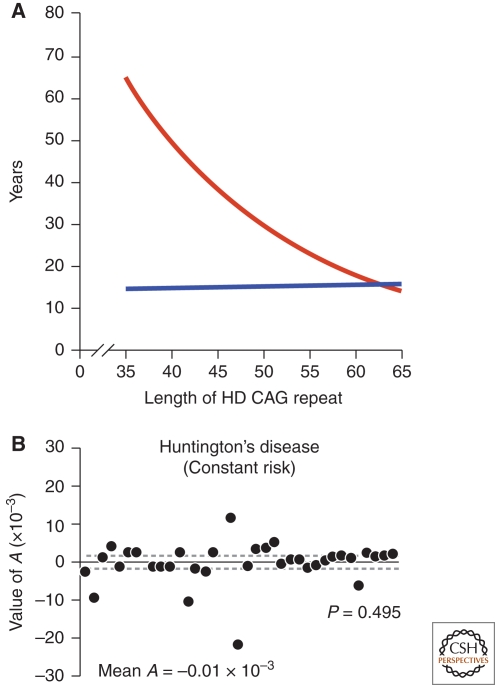
The role of the CAG expansion in IT-15 on HD pathogenesis. (A) Correlation of HD CAG-repeat length with age at onset. Best-fit curves for age at neurological onset (red) and duration of disease from onset to death (blue), plotted against CAG-repeat length for the expanded mutant allele from Huntington disease (HD) patients. Age at onset is strongly correlated with the CAG-repeat length (r2 = 0.54; p < 0.001), and duration of disease shows no correlation with the CAG-repeat length, suggesting that, after onset of HD, factors independent of the original trigger of pathogenesis determine the rate of progression. Adapted with permission from (Gusella and MacDonald 2006). (B) The kinetics of metabolic decline in Huntington's disease patients is best fit by a constant risk of cell loss. Values of A, the exponent of an equation relating longitudinal metabolic changes to time, do not differ significantly from zero (P = 0.495, Student's t-test). Each point represents the estimated A for an individual patient. Solid line, mean A (mean ± s.d.: −0.01 × 10−3 + 5.3 × 10−3 mM/h1, n = 38 patients) across patient population; dashed lines, 95% confidence interval for the mean value of A (−1.7 × 10−3, 1.7 × 10−3). Adapted with permission from (Clarke et al. 2000).
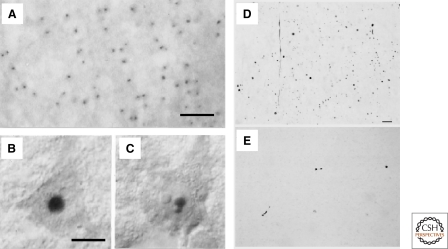
HD is characterized by abnormal protein deposits containing mutant huntingtin. (A) Huntingtin immunoreactivity in neuronal intranuclear inclusions (hNIIs) and dystrophic neurites in HD brain. Cortex of a juvenile patient shows numerous hNIIs prominently stained. (B and C) Cortical pyramidal neurons in a different juvenile patient shown with Nomarski optics contain one (B) and two (C) hNIIs. A–C, Adapted with permission from (DiFiglia et al. 1997). The nucleolus in each cell is unlabeled. (D–E) Huntingtin aggregates in human postmortem cerebral cortex and striatum from a presymptomatic case. Light micrographs are from the insular cortex (D) and dorsal striatum (E). Large numbers of EM48-immunoreactive aggregates of a wide variety of shapes and sizes are visible in cortex. All of these aggregates are in the neuropil. In contrast, striatal aggregates are exceedingly uncommon. Scale bar, 70 µm. D–E, Adapted with permission from (Gutekunst et al. 1999).
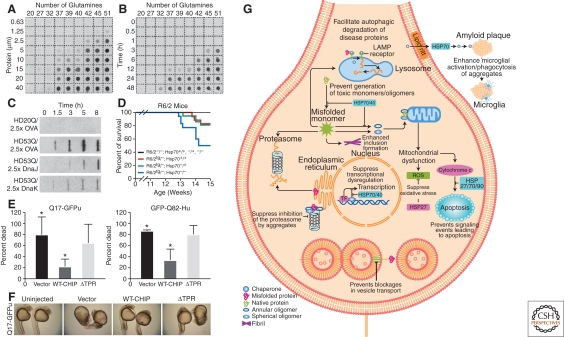
Aggregation of polyQ expansions is regulated by a network of chaperones and cochaperones. (A) Concentration dependence of polyQ-containing amino-terminal exon 1 peptide fragment of huntingtin (HDex1p) aggregation. Glutathione S-transferase (GST)-HDex1 proteins with polyQ tracts of different lengths were incubated at the indicated concentrations with trypsin for 24 h at 37°C. Aliquots (200 ng) of each protein were then diluted into 0.2 ml of 2% SDS/50 mM dithiothreitol, boiled for 3 min, and filtered through a cellulose acetate membrane. Captured aggregates were detected by incubation with antiAG51 serum (1:1000), followed by incubation with alkaline phosphatase-conjugated antirabbit secondary antibody and the fluorescent substrate AttoPhos. (B) Time course of HDex1p aggregation. The various GST-HDex1 proteins were incubated at a concentration of 20 µM with trypsin. At the indicated times, aliquots (200 ng) of each protein were removed and analyzed by the filter retardation assay as in A. A–B, Adapted with permission from (Scherzinger et al. 1999). (C) Modulation of aggregation of amino-terminal exon 1 fragment of huntingtin (HDexon 1) by Hsp70 and Hsp40 in vitro. Time-dependent formation of SDS-insoluble aggregates of HD20Q and HD53Q (3 µM) in the presence and absence of ovalbumin (OVA) and DnaJ (Hsp40) or DnaK (Hsp70) of Escherichia coli (7.5 µM each) in the absence of ATP as detected in filter-trap assays. Chaperones were added when aggregation was initiated by proteolytic cleavage of GST-HD fusion proteins. OVA served as a nonchaperone control protein. Adapted with permission from Muchowski et al. (2000). (D) Deletion of Hsp70.1 and Hsp70.3 decreases survival in the R6/2 mouse model of HD. Kaplan–Meier survival curve for the indicated genotypes [R6/2−/−; Hsp70+/+ (n = 21), R6/2tg/−; Hsp70+/+ (n = 18), R6/2−/−; Hsp70−/+ (n = 27), R6/2tg/−; Hsp70−/+ (n = 22), R6/2−/−; Hsp70−/− (n = 18), and R6/2tg/−; Hsp70−/− (n = 18)] shows that the absence of Hsp70.1/3 significantly decreased survival of R6/2 mice (log rank: p = 0.033). Nonontransgenic, Hsp70 heterozygous knock-out, or Hsp70 homozygous knock-out mice died during the 14 week time course. Adapted with permission from (Wacker et al. 2009). (E) Carboxy-terminal heat shock protein 70-interacting protein (CHIP) suppresses polyQ toxicity in developing zebrafish. Quantitation of embryo death 24 h after fertilization, after injection at the one-cell stage of Q71-GFPu (left) or GFP-Q82-Htt (right) together with the indicated CHIP plasmids. Death is decreased by coexpression of WT-CHIP but not of mutant CHIP-ΔTPR (deletion of tetratrico peptide repeat). The bars depict the mean and SD of four independent injections for Q71-GFPu (>120 total animals analyzed) and three independent injections for GFP-Q82-Htt (>80 total animals analyzed). *p<0.02, significant difference between groups. (F) Representative bright-field images of 24-h-old survivors from (E) coinjected with Q71-GFPu and the indicated empty vector, WT-CHIP, or mutant CHIP. WT-CHIP partially restores normal embryo length, optical clarity, and development of head, tail, and somite structures. (E–F) Adapted with permission from (Miller et al. 2005). (G) Direct and indirect effects of molecular chaperones on disease protein toxicity. Molecular chaperones might prevent toxicity by blocking inappropriate protein interactions, by facilitating disease protein degradation or sequestration, and by blocking downstream signaling events that lead to neuronal dysfunction and apoptosis. ER, endoplasmic reticulum; ERAD, endoplasmic reticulum-associated degradation; HSP, heat shock protein; LAMP, lysosomal-associated membrane protein; ROS, reactive oxygen species; TF, transcription factor. Adapted with permission from (Muchowski and Wacker, 2005).
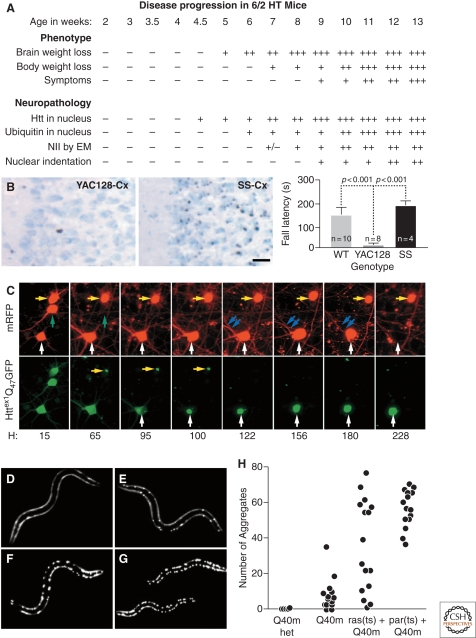
HD pathogenesis unfolds as a complex temporal dynamic, instigated by polyQ-expanded huntingtin but shaped by a network of cellular adaptive responses. (A) In the R6/2 mouse model of HD, IB formation precedes and correlates to behavioral deficits. Adapted with permission from (Davies et al. 1997). However, the Shortstop mouse model of HD (B) has more IBs (left, micrographs) but fewer behavioral deficits (right, graph) than the YAC128 mouse model of HD. Adapted with permission from (Slow et al. 2005). (C) The paradox could be explained if IB formation behaves as a coping response to more diffuse forms of mutant huntingtin. Here striatal neurons were transfected with mutant huntingtin tagged with GFP and followed longitudinally. Levels of diffuse mutant huntingtin decrease to nearly baseline levels after IB formation and, at the same, begin to show a lower risk of death (i.e., better survival). Adapted with permission from Arrasate et al. (2004). (D–H) Progressive disruption of cellular folding capacity by misfolded proteins. Fluorescent images of representative L2 larvae at the permissive temperature of heterozygous (D) or homozygous (E) Q40 m, ras (ts)+Q40 m (F), and paramyosin (ts)+Q40 m (G) strains. (H) Number of visible aggregates in L2 larvae expressing indicated proteins. ras (ts)+Q40 m in (F) and (G) denotes the fluorescent progeny of an F2 ras (ts) animal expressing Q40 m; these progeny could be either homozygous or heterozygous for Q40 m. Adapted with permission from Gidalevitz et al. (2006).
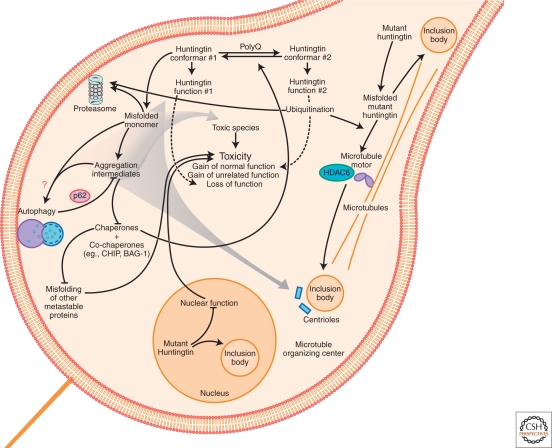
Mechanisms of neurodegeneration induced by protein malfolding in HD. A diagram depicting some of the pathways by which misfolded mutant huntingtin is collected into IBs or cleared. Toxicity could result from direct aberrant interactions between mutant huntingtin and myriad specific effector proteins. Mutant huntingtin could also cause neurodegeneration indirectly by stressing the protein homeostasis system sufficiently that other metastable proteins fail to fold properly and widespread dysfunction of the proteome ensues. Mutant huntingtin induces neurodegeneration predominantly through gain-of-function (GOF) mechanisms, although huntingtin loss-of-function (LOF) may have a role. GOF mechanisms may involve functions unrelated to huntingtin's normal function, but simultaneous GOF and LOF might also result if polyQ expansion increased a normal function of huntingtin to supraphysiological levels at the expense of another normal function of huntingtin.
References
-
- Al-Ramahi I, Lam YC, Chen H-K, de Gouyon B, Zhang M, Pérez AM, Branco J, de Haro M, Patterson C, Zoghbi HY, et al. 2006. CHIP protects from the neurotoxicity of expanded and wild-type ataxin-1 and promotes their ubiquitination and degradation. J Biol Chem 281: 26714–26724 - PubMed
-
- Ardley HC, Hung C-C, Robinson PA 2005. The aggravating role of the ubiquitin–proteasome system in neurodegeneration. FEBS Lett 579: 571–576 - PubMed
-
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S 2004. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431: 805–810 - PubMed
-
- Augood SJ, Faull RLM, Love DR, Emson PC 1996. Reduction in enkephalin and substance P messenger RNA in the striatum of early grade Huntington's disease: A detailed cellular in situ hybridization study. Neuroscience 72: 1023–1036 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical