

Endoplasmic-reticulum calcium depletion and disease
- PMID: 21441595
- PMCID: PMC3098671
- DOI: 10.1101/cshperspect.a004317
Endoplasmic-reticulum calcium depletion and disease
Abstract
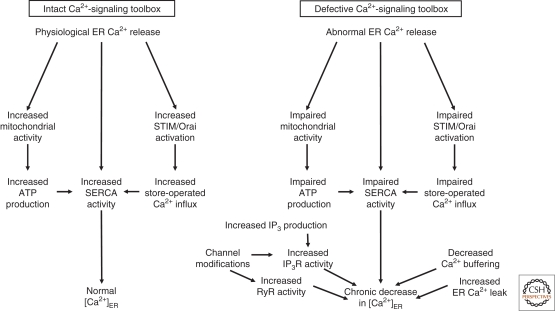
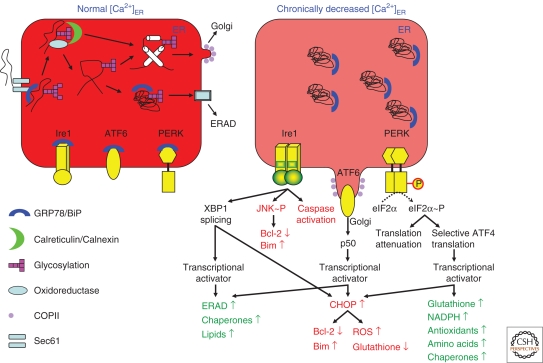
The endoplasmic reticulum (ER) as an intracellular Ca(2+) store not only sets up cytosolic Ca(2+) signals, but, among other functions, also assembles and folds newly synthesized proteins. Alterations in ER homeostasis, including severe Ca(2+) depletion, are an upstream event in the pathophysiology of many diseases. On the one hand, insufficient release of activator Ca(2+) may no longer sustain essential cell functions. On the other hand, loss of luminal Ca(2+) causes ER stress and activates an unfolded protein response, which, depending on the duration and severity of the stress, can reestablish normal ER function or lead to cell death. We will review these various diseases by mainly focusing on the mechanisms that cause ER Ca(2+) depletion.
Figures
References
-
- Abdelrahim M, Newman K, Vanderlaag K, Samudio I, Safe S 2006. 3,3'-diindolylmethane (DIM) and its derivatives induce apoptosis in pancreatic cancer cells through endoplasmic reticulum stress-dependent upregulation of DR5. Carcinogenesis 27: 717–728 - PubMed
-
- Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schoneich C, Cohen RA 2004. S-glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med 10: 1200–1207 - PubMed
-
- Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM 2005. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res 97: 1314–1322 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous